Całkowite zanieczyszczenie gleby charakteryzuje całkowitą ilość metali ciężkich. O dostępności elementów dla roślin decydują ich mobilne formy. Dlatego zawartość mobilnych form metali ciężkich w glebie jest najważniejszym wskaźnikiem charakteryzującym sytuację sanitarno-higieniczną i determinuje konieczność podjęcia działań rekultywacyjnych w zakresie detoksykacji.
W zależności od użytego ekstrahenta ekstrahowana jest różna ilość ruchomej postaci metalu ciężkiego, która zgodnie z pewną konwencją może być uznana za dostępną dla roślin. Do ekstrakcji ruchomych form metali ciężkich stosuje się różne związki chemiczne o nierównych właściwościach ekstrakcyjnych: kwasy, sole, roztwory buforowe i wodę. Najpopularniejszymi ekstrahentami są 1N HCl i bufor octanu amonu o pH 4,8. Obecnie nie zgromadzono wystarczającej ilości materiału doświadczalnego, aby scharakteryzować zależność zawartości metali ciężkich w roślinach, które są ekstrahowane różnymi roztworami chemicznymi, od ich stężenia w glebie. Złożoność tej sytuacji wynika również z faktu, że dostępność mobilnej formy metalu ciężkiego dla roślin zależy w dużej mierze od właściwości gleby i specyfiki roślin. Jednocześnie zachowanie każdego elementu w glebie ma swoje specyficzne wzorce.
W celu zbadania wpływu właściwości gleb na przemiany związków metali ciężkich przeprowadzono eksperymenty modelowe z glebami o wyraźnie odmiennych właściwościach (tab. 8). Użytymi ekstrahentami były mocny kwas, 1N HNO3, obojętna sól Ca(NO3)2, roztwór buforowy octanu amonu i woda.

Wskazują na to dane analityczne podane w tabelach 9-12. że zawartość rozpuszczalnych w kwasach związków cynku, ołowiu i kadmu, przechodzących do ekstraktu 1n HNO3, jest zbliżona do ich ilości wprowadzonej do gleby.Ekstraktant ten wyekstrahował 78-90% Pb, 88-100% Cd i 78- 96% Zn, który dostał się do gleby . Ilość trwale związanych związków tych pierwiastków zależała od poziomu żyzności gleby. Ich zawartość w słabo uprawianej glebie bielicowo-leśnej była niższa niż w uprawianej glebie bielicowo-leśnej i typowym czarnoziemie.
Ilość związków wymiennych Cd, Pb i Zn wyekstrahowanych 1-n roztworem soli obojętnej Ca(NO3)2 była kilkakrotnie mniejsza od ich masy wprowadzonej do gleby, a także zależała od poziomu żyzności gleby. Najmniej treści pierwiastki wyekstrahowane roztworem Ca(NO3)2 otrzymano na czarnoziemie. Wraz ze wzrostem uprawy gleby bielicowo-błękitnej zmniejszyła się również mobilność metali ciężkich. Sądząc po ekstrakcie soli, związki kadmu są najbardziej ruchliwe, a związki cynku są nieco mniej ruchliwe. Najmniejszą ruchliwością charakteryzowały się związki ołowiu wyekstrahowane solą obojętną.
O zawartości mobilnych form metali ekstrahowanych roztworem buforowym octanu amonu o pH 4,8 decydował również przede wszystkim rodzaj gleby, jej skład oraz właściwości fizykochemiczne.
W przypadku wymiennych (ekstrahowalnych 1 N Ca(NO3)2) form tych pierwiastków zachowana jest prawidłowość, która wyraża się wzrostem ilości mobilnych związków Cd, Pb i Zn w glebie kwaśnej oraz ruchliwością Cd i Zn są wyższe niż Pb. Ilość kadmu wyekstrahowanego tym ekstraktem wynosiła 90–96% zastosowanej dawki dla gleby słabo uprawianej, 70–76% dla gleby uprawianej na podłożu sodowo-bielicowym, a 44–48% dla czarnoziemu. Ilości cynku i ołowiu przechodzące do roztworu buforowego CH3COONH4 są równe odpowiednio: 57-71 i 42-67% dla gleby sodowo-bielicowej słabo uprawianej, 49-70 i 37-48% dla gleby uprawianej średnio; 46-65 i 20-42% dla czarnoziemu. Spadek zdolności ekstrakcji CH3COONH4 dla ołowiu na czarnoziemie można wytłumaczyć powstawaniem jego bardziej stabilnych kompleksów i związków ze stabilnymi związkami humusowymi.
Gleby użyte w doświadczeniu modelowym różniły się pod wieloma względami. żyzność gleby, ale w największym stopniu w charakterystyce kwasowej i liczbie wymiennych zasad. Dostępne w literaturze i uzyskane przez nas dane doświadczalne wskazują, że reakcja ośrodka w glebie silnie wpływa na ruchliwość pierwiastków.
Wzrost stężenia jonów wodorowych w roztworze glebowym prowadził do przejścia słabo rozpuszczalnych soli ołowiu w bardziej rozpuszczalne sole (szczególnie charakterystyczna jest przemiana PbCO3 w Pb (HCO3) 2 (B.V. Nekrasov, 1974). Ponadto zakwaszenie zmniejsza stabilność kompleksów ołowiowo-próchniczych.Wartość pH roztworu glebowego jest jednym z najważniejszych parametrów decydujących o ilości sorpcji jonów metali ciężkich przez glebę.Wraz ze spadkiem pH wzrasta rozpuszczalność większości metali ciężkich i w konsekwencji ich mobilność w układzie gleba faza stała – roztwór J. Esser, N. Bassam (1981 ) badając mobilność kadmu w tlenowych warunkach glebowych stwierdzili, że w zakresie pH 4-6 mobilność kadmu zależy od siły jonowej roztworu, przy pH powyżej 6 pierwszorzędne znaczenie nabiera sorpcja tlenków manganu. Rozpuszczalne związki organiczne, zdaniem autorów, tworzą tylko słabe kompleksy z kadmem i wpływają na jego sorpcję dopiero przy pH 8.
Najbardziej ruchliwą i dostępną dla roślin częścią związków metali ciężkich w glebie jest ich zawartość w roztworze glebowym. Ilość jonów metali przedostających się do roztworu glebowego określa toksyczność danego pierwiastka w glebie. Stan równowagi w układzie faza stała-roztwór determinuje procesy sorpcyjne, których charakter i kierunek zależą od właściwości i składu gleby. Wpływ właściwości gleby na mobilność metali ciężkich i ich przechodzenie do ekstraktu wodnego potwierdzają dane dotyczące inna kwota rozpuszczalne w wodzie związki Zn, Pb i Cd przenoszone z gleb o różnym poziomie żyzności przy tych samych dawkach zastosowanych metali (tab. 13). W porównaniu z czarnoziemem w glebie ornatowo-bielicowej było więcej rozpuszczalnych w wodzie związków metali. Największą zawartość rozpuszczalnych w wodzie związków Zn, Pb i Cd stwierdzono w glebie słabo uprawianej. Uprawa gleby zmniejszyła mobilność metali ciężkich. W glebie bagienno-bielicowej słabo uprawianej zawartość rozpuszczalnych w wodzie form Zn. Pb i Cd były o 20-35% wyższe niż w glebie uprawianej średnio i 1,5-2,0 razy wyższe niż w typowym czarnoziemie. Wzrost żyzności gleby, któremu towarzyszy wzrost zawartości próchnicy, fosforanów, neutralizacja nadmiernej kwasowości oraz wzrost właściwości buforowych, prowadzi do zmniejszenia zawartości najbardziej agresywnej, rozpuszczalnej w wodzie formy metali ciężkich.
Decydującą rolę w rozmieszczeniu metali ciężkich w układzie gleba-roztwór odgrywają procesy sorpcji-desorpcji na fazie stałej gleby, które determinowane są właściwościami gleby i nie zależą od postaci wprowadzony związek. Powstałe związki metali ciężkich z fazą stałą gleby są bardziej stabilne termodynamicznie niż wprowadzone związki i determinują stężenie pierwiastków w roztworze glebowym (R.I. Pervunina, 1983).
Gleba jest silnym i aktywnym pochłaniaczem metali ciężkich, jest w stanie mocno wiązać i tym samym ograniczać dopływ toksyn do roślin. Składniki mineralne i organiczne gleby aktywnie dezaktywują związki metali, ale ilościowe przejawy ich działania zależą od rodzaju gleby (BA. Bolshakov i in., 1978, V. B. Ilyin, 1987).
Zgromadzony materiał doświadczalny wskazuje na to. że największą ilość metali ciężkich wydobywa z gleby 1 n kwaśny ekstrakt. Jednocześnie dane są zbliżone do całkowitej zawartości pierwiastków w glebie. Tę formę elementów można uznać za całkowitą zapasową ilość zdolną do przejścia w mobilną formę mobilną. Zawartość metali ciężkich po wyekstrahowaniu z gleby buforem octanowo-amonowym charakteryzuje bardziej ruchomą część. Jeszcze bardziej mobilna jest forma wymiany metalu ciężkiego. ekstrahowany obojętnym roztworem soli fizjologicznej. VS. Gorbatow i N.G. Zyrin (1987) uważają, że najbardziej dostępna dla roślin jest forma wymienna metali ciężkich, selektywnie ekstrahowana roztworami soli, której anion nie tworzy kompleksów z metalami ciężkimi, a kation ma dużą siłę wypierania. To właśnie te właściwości posiada Ca(NO3)2 użyty w naszym eksperymencie. Najbardziej agresywne rozpuszczalniki - kwasy, najczęściej stosowane 1N HCl i 1N HNO3, wydobywają z gleby nie tylko przyswajalne przez rośliny formy, ale także część pierwiastka brutto, które stanowią najbliższą rezerwę do przejścia w związki mobilne.
Stężenie w roztworze glebowym metali ciężkich wyekstrahowanych ekstraktem wodnym charakteryzuje najbardziej aktywną część ich związków. Jest to najbardziej agresywna i dynamiczna frakcja metali ciężkich, która charakteryzuje stopień ruchliwości pierwiastków w glebie. Wysoka zawartość rozpuszczalnych w wodzie form TM może prowadzić nie tylko do skażenia produktów roślinnych, ale także do gwałtownego spadku plonu aż do jego śmierci. Przy bardzo wysokiej zawartości rozpuszczalnej w wodzie formy metali ciężkich w glebie staje się niezależnym czynnikiem determinującym wielkość plonu i stopień jego zanieczyszczenia.
W naszym kraju zgromadzono informacje o zawartości mobilnej formy TM w nieskażonych glebach, głównie tych, które są znane jako pierwiastki śladowe - Mn, Zn, Cu, Mo. Co (Tabela 14). Do określenia formy mobilnej najczęściej stosowano ekstrahenty indywidualne (wg Peive Ya.V. i Rinkis G.Ya.). Jak widać z tabeli 14, gleby poszczególnych rejonów różniły się istotnie ilością ruchomej postaci tego samego metalu.
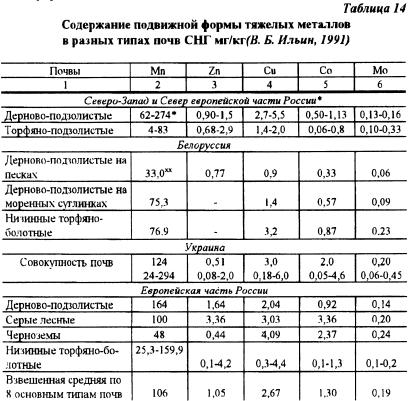
Powodem może być, według V.B. Ilyin (1991), cechy genetyczne gleb, przede wszystkim specyfika składu granulometrycznego i mineralogicznego, poziom próchnicy, odczyn środowiska. Z tego powodu gleby jednego regionu naturalnego mogą się znacznie różnić, a ponadto mogą mieć ten sam typ genetyczny w tym regionie.
Różnica między minimalną i maksymalną ilością napotkanej formy ruchomej może mieścić się w porządku matematycznym. Nie ma absolutnie niewystarczającej informacji na temat zawartości w glebach formy mobilnej Pb, Cd, Cr, Hg i innych najbardziej toksycznych pierwiastków. Prawidłowa ocena ruchliwości TM w glebie utrudnia stosowanie jako ekstrahentów chemikaliów, które znacznie różnią się pod względem siły rozpuszczania. I tak np. 1 N HCl wyekstrahowano formy ruchome z poziomu strugowego w mg/kg: Mn - 414, Zn - 7,8 Ni - 8,3, Cu - 3,5, Pb - 6,8, Co - 5,3 (gleba Zachodnia Syberia), podczas gdy odzyskano 2,5% CH3COOH 76; 0,8; 1.2; 1.3; 0,3; 0,7 (gleby obwodu Tomsk Ob, dane z Ilyin, 1991). Materiały te wskazują, że 1 N HCl wyekstrahowany z gleby, z wyjątkiem cynku, około 30% metali w całkowitej ilości, a 2,5% CH3COOH - mniej niż 10%. Dlatego ekstrahent 1N HCl, szeroko stosowany w badaniach agrochemicznych i charakterystyce gleby, ma wysoką zdolność mobilizacji rezerw metali ciężkich.
Główna część mobilnych związków metali ciężkich jest ograniczona do próchnicy lub zakorzenionych poziomów gleby, w których aktywnie zachodzą procesy biochemiczne i zawierają wiele substancji organicznych. Metale ciężkie. wchodzące w skład kompleksów organicznych mają dużą mobilność. V.B. Ilyin (1991) wskazuje na możliwość akumulacji metali ciężkich w poziomach iluwialnych i węglanowych, do których drobne cząstki nasycone metalami ciężkimi migrują z warstwy przykrywającej i rozpuszczalnych w wodzie form pierwiastków. W poziomach iluwialnych i węglanowych wytrącają się związki zawierające metal. Najbardziej ułatwia to gwałtowny wzrost pH pożywki w glebie tych poziomów, spowodowany obecnością węglanów.
Zdolność metali ciężkich do akumulacji w niższych warstwach glebowych dobrze obrazują dane dotyczące profili glebowych na Syberii (tab. 15). W horyzoncie próchniczym obserwuje się podwyższoną zawartość wielu pierwiastków (Sr, Mn, Zn, Ni itp.) niezależnie od ich genezy. W wielu przypadkach wyraźnie widać wzrost zawartości ruchomego Sr w horyzoncie węglanowym. Ogólna zawartość form mobilnych w mniejszej ilości jest typowa dla gleb piaszczystych, znacznie więcej dla gliniastych. Oznacza to, że istnieje ścisły związek między zawartością ruchomych form pierwiastków a składem granulometrycznym gleb. Podobną pozytywną zależność można prześledzić między zawartością ruchomych form metali ciężkich a zawartością próchnicy.
Zawartość mobilnych form metali ciężkich podlega silnym wahaniom, co związane jest ze zmienną aktywnością biologiczną gleb oraz wpływem roślin. Tak więc według badań przeprowadzonych przez V.B. Iljin zawartość mobilnego molibdenu w glebie bielicowej i południowo-czarnoziemnej w okresie wegetacji zmieniała się 5 razy.
W ostatnich latach w niektórych jednostkach naukowych badano wpływ wieloletniego stosowania nawozów mineralnych, organicznych i wapniowych na zawartość mobilnych form metali ciężkich w glebie.
W agrochemicznej stacji doświadczalnej Dolgoprudnaya (DAOS, obwód moskiewski) przeprowadzono badania akumulacji metali ciężkich, pierwiastków toksycznych w glebie oraz ich mobilności w warunkach długotrwałego stosowania nawozów fosforowych na glebie wapienno-bielicowej ciężkiej gliniastej (Yu.A. Potatueva i in., 1994. ). Systematyczne stosowanie nawozów balastowych i skoncentrowanych przez 60 lat, różnych form fosforanów przez 20 lat oraz fosforytów z różnych złóż przez 8 lat nie miało istotnego wpływu na całkowitą zawartość metali ciężkich i pierwiastków toksycznych (TE) w gleba, ale doprowadziła do wzrostu mobilności, zawiera trochę TM i TE. Zawartość form mobilnych i rozpuszczalnych w wodzie w glebie wzrosła około 2-krotnie przy systematycznym stosowaniu wszystkich badanych form nawozów fosforowych, jednak wyniosła zaledwie 1/3 MPC. Ilość strontu mobilnego wzrosła 4,5 razy w glebie, która otrzymała superfosfat prosty. Wprowadzenie surowych fosforytów ze złoża Kingisep spowodowało wzrost zawartości form mobilnych w glebie (AAB pH 4,8): ołowiu o 2 razy, niklu o 20% i chromu o 17%, co wyniosło 1/4 i 1/10 członków RPP. W glebie, do której wprowadzono surowe fosforyty ze złoża Chilisai, odnotowano wzrost zawartości chromu mobilnego o 17% (tab. 16).
Porównanie danych eksperymentalnych wieloletnich doświadczeń polowych z DAOS z normami sanitarno-higienicznymi na zawartość mobilnych form metali ciężkich w glebie, a przy ich braku z zaleceniami proponowanymi w literaturze wskazuje, że zawartość form mobilnych tych pierwiastków w glebie było poniżej dopuszczalnych poziomów. Te dane doświadczalne wskazują, że nawet bardzo długotrwałe stosowanie nawozów fosforowych przez 60 lat nie doprowadziło do przekroczenia poziomu MPC w glebie, ani w postaci brutto, ani w ruchliwych formach metali ciężkich. Jednocześnie dane te wskazują, że reglamentacja metali ciężkich w glebie jedynie formami zgrubnymi nie jest dostatecznie uzasadniona i powinna być uzupełniona o zawartość formy mobilnej, która odzwierciedla zarówno właściwości chemiczne samych metali, jak i właściwości gleby, na której rosną rośliny.
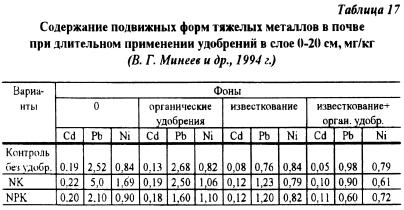
Na podstawie wieloletniego doświadczenia terenowego, opracowanego pod kierunkiem akademika N.S. Avdonin w bazie doświadczalnej Moskiewskiego Uniwersytetu Państwowego „Czasznikowo” przeprowadzono badanie wpływu długotrwałego stosowania nawozów mineralnych, organicznych, wapniowych i ich kombinacji na zawartość mobilnych form metali ciężkich w glebie przez 41 lat (V.G. Mineev i in., 1994). Wyniki badań w tabeli 17 wykazały, że stworzenie optymalnych warunków do wzrostu i rozwoju roślin istotnie obniżyło zawartość mobilnych form ołowiu i kadmu w glebie. Systematyczne stosowanie nawozów azotowo-potasowych, zakwaszające roztwór glebowy i zmniejszające zawartość mobilnego fosforu, podwoiło stężenie mobilnych związków ołowiu i niklu oraz 1,5-krotnie zwiększyło zawartość kadmu w glebie.
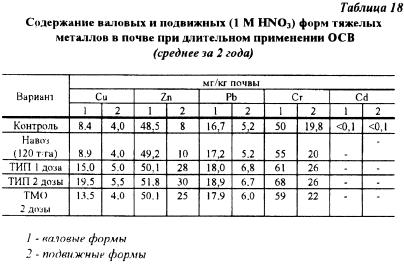
W trakcie wieloletniego użytkowania komunalnych osadów ściekowych badano zawartość ogólnych i mobilnych form TM w glebie bielicowo-bielcowo-gliniastej Białorusi: termofilnie fermentowanych z pól namułowych (TIP) i termofilnie fermentowanych z późniejszym odwadnianiem mechanicznym (TMD).
W ciągu 8 lat badań nasycenie płodozmianu OCB wyniosło 6,25 t/ha (pojedyncza dawka) i 12,5 t/ha (podwójna dawka), czyli około 2-3 razy więcej niż dawki zalecane.
Jak widać z Tabeli 18, widać wyraźny wzorzec wzrostu zawartości brutto i mobilnych form TM w wyniku trzykrotnego zastosowania WWS. Ponadto największą ruchliwością charakteryzuje się cynk, którego ilość w postaci mobilnej wzrosła 3-4-krotnie w porównaniu z glebą kontrolną (N.P. Reshetsky, 1994). Jednocześnie zawartość związków mobilnych kadmu, miedzi, ołowiu i chromu nie uległa istotnym zmianom.
Badania naukowców strony białoruskiej - x. uczelnie wykazały, że przy wprowadzaniu osadów ściekowych (mokry osad z pól namułowych, SIP, TMF) zauważalny był wzrost zawartości mobilnych form pierwiastków w glebie, ale najsilniej kadmu, cynku i miedzi (tab. 19). . Wapnowanie praktycznie nie miało wpływu na mobilność metali. Według autorów. zastosowanie ekstraktu w 1 N HNO3 do scharakteryzowania stopnia ruchliwości metali nie jest skuteczne, ponieważ przechodzi do niego ponad 80% całkowitej zawartości pierwiastka (A.I. Gorbyleva i in., 1994).

Ustalenie pewnych zależności zmian ruchliwości TM w glebie od poziomu kwasowości przeprowadzono w doświadczeniach mikropolowych na ługowanych czarnoziemach Centralnego Czarnoziemu Federacji Rosyjskiej. Jednocześnie oznaczano kadm, cynk i ołów w ekstraktach: kwas solny, azotowy, siarkowy, bufor octanu amonu o pH 4,8 i pH 3,5, saletra amonowa, woda destylowana. Stwierdzono ścisły związek między całkowitą zawartością cynku a jego formami mobilnymi wyekstrahowanymi kwasami R=0,924-0,948. Przy stosowaniu AAB pH 4,8 R=0,784, AAB pH 3,5=0,721. Ołów odzyskany przez kwas solny i azotowy słabiej skorelowany z zawartością brutto: R=0,64-0,66. Pozostałe ekstrakty miały znacznie niższe wartości współczynników korelacji. Korelacja między kwasowymi związkami kadmu ekstrahowanych a rezerwami brutto była bardzo wysoka (R=0,98-0,99). przy ekstrakcji AAB pH 4,8-R=0,92. Użycie innych ekstraktów dało wyniki wskazujące na słabą zależność między ogólnymi i ruchomymi formami metali ciężkich w glebie (N.P. Bogomazov, P.G. Akulov, 1994).
W wieloletnim eksperymencie polowym (Ogólnorosyjski Instytut Badawczy Lnu, Region Twerski) przy długotrwałym stosowaniu nawozów na glebie bielicowo-błękitnej, udział ruchomych związków metali z zawartości ich potencjalnie dostępnych form zmniejszył się szczególnie wyraźnie w 3 rok od następstw wapna w dawce 2 g q. (tab. 20). W 13 roku od następstw wapno w tej samej dawce redukowało jedynie zawartość mobilnego żelaza i glinu w glebie. w 15 roku - żelazo, aluminium i mangan (L.I. Petrova. 1994).

Dlatego w celu zmniejszenia zawartości mobilnych form ołowiu i miedzi w glebie konieczne jest wielokrotne wapnowanie gleb.
Badania ruchliwości metali ciężkich w czarnoziemach regionu Rostowa wykazały, że w warstwie metrowej zwykłych czarnoziemów ilość cynku wyekstrahowanego ekstraktem buforowym octanu amonu o pH 4,8 wahała się w granicach 0,26-0,54 mg/kg. mangan 23,1-35,7 mg / kg, miedź 0,24-0,42 (G.V. Agafonov, 1994), Porównanie tych liczb z brutto rezerwami pierwiastków śladowych w glebie tych samych działek wykazało, że ruchliwość różne elementy różni się znacznie. Cynk na czarnoziemie węglanu jest 2,5-4,0 razy mniej dostępny dla roślin niż miedź i 5-8 razy mniej niż mangan (Tabela 21).

Tak więc pokazują wyniki przeprowadzonych badań. że problem mobilności metali ciężkich w glebie jest złożony i wieloczynnikowy. Zawartość mobilnych form metali ciężkich w glebie zależy od wielu warunków. Główna recepcja prowadzącym do obniżenia zawartości tej formy metali ciężkich jest wzrost żyzności gleby (wapnowanie, wzrost zawartości próchnicy i fosforu itp.). Jednocześnie nie ma ogólnie przyjętej formuły dla metali ruchomych. W tej sekcji zaproponowaliśmy nasze zrozumienie różnych frakcji metali ruchomych w glebie:
1) całkowity zasób form mobilnych (wyekstrahowanych kwasami);
2) mobilna forma mobilna (odzyskiwalna za pomocą roztworów buforowych):
3) wymienne (wyekstrahowane solami obojętnymi);
4) rozpuszczalny w wodzie.
ZAWARTOŚĆ
Wstęp
1. Pokrywa gleby i jej zastosowanie
2. Erozja gleby (wodna i wiatrowa) i sposoby radzenia sobie z nią
3. zanieczyszczenia przemysłowe gleba
3.1 Kwaśny deszcz
3.2 Metale ciężkie
3.3 Zatrucie ołowiem
4. Higiena gleby. Utylizacja odpadów
4.1 Rola gleby w metabolizmie
4.2 Ekologiczny związek między glebą a wodą i odpadami płynnymi (ściekami)
4.3 Limity obciążenia gleby odpadami stałymi (odpady z gospodarstw domowych i ulic, odpady przemysłowe, suche osady po sedymentacji ścieków, substancje promieniotwórcze)
4.4 Rola gleby w rozprzestrzenianiu się różnych chorób
4.5 Szkodliwe skutki głównych rodzajów zanieczyszczeń (odpady stałe i płynne) prowadzące do degradacji gleby
4.5.1 Dekontaminacja nieczystości płynnych w glebie
4.5.2.1 Dekontaminacja odpadów stałych w glebie
4.5.2.2 Zbieranie i usuwanie odpadów
4.5.3 Ostateczne usuwanie i usuwanie
4.6 Utylizacja odpadów promieniotwórczych
Wniosek
Lista wykorzystanych źródeł
Wstęp.
Pewna część gleb, zarówno w Rosji, jak i na całym świecie, co roku wychodzi z obiegu rolniczego z różnych powodów, które są szczegółowo omówione w UIR. Tysiące lub więcej hektarów ziemi jest dotkniętych erozją, kwaśnymi deszczami, złym zarządzaniem i toksycznymi odpadami. Aby tego uniknąć, należy zapoznać się z najbardziej produktywnymi i niedrogimi metodami rekultywacji gruntów (patrz definicja rekultywacji w głównej części pracy), które zwiększają żyzność pokrywy glebowej, a przede wszystkim z negatywnymi wpływ na samą glebę i jak jej uniknąć.
Badania te dostarczają wglądu w szkodliwy wpływ na glebę i zostały przeprowadzone w wielu książkach, artykułach i czasopismach naukowych dotyczących zagadnień glebowych i środowiskowych.
Sam problem zanieczyszczenia i degradacji gleby zawsze był aktualny. Teraz możemy dodać do tego, co zostało powiedziane, że w naszych czasach wpływ antropogeniczny bardzo wpływa na przyrodę i tylko rośnie, a gleba jest dla nas jednym z głównych źródeł pożywienia i odzieży, nie mówiąc już o tym, że po niej chodzimy i zawsze będzie z nią w bliskim kontakcie.
1. Pokrywa glebowa i jej zastosowanie.
Pokrywa glebowa jest najważniejszą formacją naturalną. O jego znaczeniu dla życia społeczeństwa decyduje fakt, że gleba jest głównym źródłem pożywienia, dostarczając 97-98% zasobów żywnościowych ludności świata. Jednocześnie pokrywa glebowa jest miejscem działalności człowieka, w którym odbywa się produkcja przemysłowa i rolna.
Podkreślając szczególną rolę żywności w życiu społeczeństwa, nawet V. I. Lenin wskazał: „Prawdziwym fundamentem gospodarki jest fundusz żywnościowy”.
Najważniejszą właściwością pokrywy glebowej jest jej żyzność, rozumiana jako całość właściwości gleby zapewniających plon upraw rolnych. Naturalną żyzność gleby reguluje inwentarz składniki odżywcze w glebie i jej reżimach wodnych, powietrznych i termicznych. Rola pokrywy glebowej w produktywności lądowych systemów ekologicznych jest duża, gdyż gleba odżywia rośliny lądowe wodą i wieloma związkami oraz jest niezbędnym składnikiem aktywności fotosyntetycznej roślin. Żyzność gleby zależy również od ilości zgromadzonej w niej energii słonecznej. Żywe organizmy, rośliny i zwierzęta zamieszkujące Ziemię gromadzą energię słoneczną w postaci fito- lub zoomass. Produktywność ziemskich systemów ekologicznych zależy od bilansu cieplnego i wodnego powierzchni Ziemi, który determinuje różnorodność form wymiany materii i substancji w obrębie geograficznej otoczki planety.
Analizując znaczenie ziemi dla produkcji społecznej, K. Marks wyróżnił dwa pojęcia: ziemia-materia i ziemia-kapitał. Należy zrozumieć pierwsze z nich ziemia, która powstała w procesie swego ewolucyjnego rozwoju oprócz woli i świadomości ludzi i jest miejscem osiedlenia się człowieka i źródłem jego pożywienia. Od momentu, gdy ziemia w procesie rozwoju społeczeństwa ludzkiego staje się środkiem produkcji, działa w nowej jakości – kapitale, bez którego proces pracy jest nie do pomyślenia, „...bo daje robotnikowi... miejsce, na którym stoi… i jego zakres procesowy…”. Z tego powodu ziemia jest uniwersalnym czynnikiem każdej ludzkiej działalności.
Rola i miejsce ziemi nie są takie same w różne pola produkcja materiałowa, zwłaszcza w przemyśle i rolnictwie. W przemyśle wytwórczym, budownictwie, transporcie ziemia jest miejscem, w którym zachodzą procesy pracy, niezależnie od naturalnej żyzności gleby. W innym charakterze jest ziemia w rolnictwie. Pod wpływem pracy ludzkiej naturalna płodność przekształca się z potencjalnej w ekonomiczną. Specyfika użytkowania zasobów ziemi w rolnictwie powoduje, że działają one w dwóch różnych jakościach, jako przedmiot pracy i środek produkcji. K. Marks zauważył: „Tylko przez nową inwestycję kapitału w działki ziemi […] ludzie powiększyli kapitał ziemski bez zwiększania materii ziemi, tj. przestrzeni ziemi”.
Ziemia w rolnictwie działa jako siła produkcyjna ze względu na swoją naturalną żyzność, która nie jest stała. Przy racjonalnym użytkowaniu ziemi, żyzność tę można zwiększyć poprzez poprawę jej reżimu wodnego, powietrznego i termicznego poprzez działania rekultywacyjne i zwiększenie zawartości składników pokarmowych w glebie. Wręcz przeciwnie, przy nieracjonalnym wykorzystaniu zasobów ziemi ich żyzność spada, w wyniku czego następuje spadek plonów. W niektórych miejscach uprawa roślin staje się całkowicie niemożliwa, zwłaszcza na glebach zasolonych i zerodowanych.
Przy niskim poziomie rozwoju sił wytwórczych społeczeństwa ekspansja produkcji żywności następuje z powodu zaangażowania nowych ziem w rolnictwo, co odpowiada ekstensywnemu rozwojowi rolnictwa. Przyczyniają się do tego dwa warunki: dostępność wolnej ziemi oraz możliwość prowadzenia działalności rolniczej na przystępnym średnim poziomie kosztów kapitałowych na jednostkę powierzchni. Takie wykorzystanie zasobów ziemi i rolnictwa jest typowe dla wielu krajów rozwijających się we współczesnym świecie.
W dobie rewolucji naukowo-technicznej nastąpiło ostre rozgraniczenie systemu rolnictwa w krajach uprzemysłowionych i rozwijających się. Te pierwsze charakteryzują się intensyfikacją rolnictwa wykorzystującą zdobycze rewolucji naukowo-technicznej, w której rolnictwo rozwija się nie dzięki zwiększeniu powierzchni gruntów uprawnych, ale dzięki zwiększeniu ilości zainwestowanego w ziemię kapitału. Dobrze znane ograniczone zasoby ziemi w większości uprzemysłowionych krajów kapitalistycznych, wzrost popytu na produkty rolne na całym świecie ze względu na wysokie tempo wzrostu liczby ludności oraz wyższy standard rolnictwa przyczyniły się do przeniesienia rolnictwa w tych krajach z powrotem w latach 50. do ścieżka intensywnego rozwoju. Przyspieszenie procesu intensyfikacji rolnictwa w uprzemysłowionych krajach kapitalistycznych związane jest nie tylko z osiągnięciami rewolucji naukowo-technicznej, ale przede wszystkim z opłacalnością lokowania kapitału w rolnictwie, które skoncentrowało produkcję rolną w rękach wielkich właścicieli ziemskich i zrujnowany drobnych rolników.
Inaczej rozwijało się rolnictwo w krajach rozwijających się. Wśród dotkliwych problemów zasobów naturalnych tych krajów można wyróżnić: niską kulturę rolniczą, która powodowała degradację gleb (wzrost erozji, zasolenia, zmniejszenie żyzności) oraz naturalną roślinność (np. lasy tropikalne), wyczerpywanie się zasobów wodnych, pustynnienie ziem, co szczególnie wyraźnie przejawia się na kontynencie afrykańskim. Wszystkie te czynniki związane z problemami społeczno-gospodarczymi krajów rozwijających się doprowadziły do chronicznych niedoborów żywności w tych krajach. Tak więc na początku lat osiemdziesiątych kraje rozwijające się kilkakrotnie ustępowały pod względem podaży zboża (222 kg) i mięsa (14 kg) na osobę odpowiednio rozwiniętym przemysłowo krajom kapitalistycznym. Rozwiązanie problemu żywnościowego w krajach rozwijających się jest nie do pomyślenia bez poważnych przemian społeczno-gospodarczych.
W naszym kraju podstawą stosunków ziemskich jest ogólnokrajowa (ogólnokrajowa) własność ziemi, która powstała w wyniku nacjonalizacji wszystkich gruntów. Stosunki agrarne budowane są na podstawie planów, zgodnie z którymi rolnictwo powinno rozwijać się w przyszłości, przy wsparciu finansowym i kredytowym państwa oraz dostarczeniu niezbędnej ilości maszyn i nawozów. Wynagrodzenie robotników rolnych według ilości i jakości pracy stymuluje stały wzrost ich standardu życia.
Całość wykorzystania funduszu ziemi odbywa się na podstawie wieloletnich planów państwowych. Przykładem takich planów było zagospodarowanie dziewiczych i odłogów na wschodzie kraju (połowa lat 50. XX w.), dzięki którym w krótkim czasie stało się możliwe wprowadzenie ponad 41 mln ha nowych terenów pod grunty orne. Innym przykładem jest zespół działań związanych z realizacją Programu Żywnościowego, który przewiduje przyspieszenie rozwoju produkcji rolnej poprzez wzrost kultury rolnictwa, szeroką realizację działań rekultywacyjnych, a także realizację działań rekultywacyjnych. szeroki program odbudowy społeczno-gospodarczej obszarów rolniczych.
Zasoby ziemi na całym świecie dostarczają pożywienia większej liczbie ludzi niż jest to obecnie dostępne i będzie w najbliższej przyszłości. Jednak ze względu na wzrost liczby ludności, zwłaszcza w krajach rozwijających się, ilość gruntów ornych na mieszkańca spada.
PODZIAŁ STRONY-- metale ciężkie, który charakteryzuje szeroką grupę zanieczyszczeń, w ostatnim czasie stał się powszechny. W różnych pracach naukowych i stosowanych autorzy w różny sposób interpretują znaczenie tego pojęcia. W związku z tym liczba pierwiastków przypisanych do grupy metali ciężkich zmienia się w szerokim zakresie. Jako kryteria przynależności stosuje się liczne cechy: masa atomowa, gęstość, toksyczność, występowanie w środowisku naturalnym, stopień zaangażowania w cykle naturalne i technogeniczne. W niektórych przypadkach definicja metali ciężkich obejmuje pierwiastki, które są kruche (na przykład bizmut) lub metaloidy (na przykład arsen).
W pracach poświęconych problematyce zanieczyszczenia środowiska” środowisko naturalne i monitoring środowiska, dziś do metale ciężkie obejmują ponad 40 metali układu okresowego D.I. Mendelejew o masie atomowej większej niż 50 jednostek atomowych: V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Mo, Cd, Sn, Hg, Pb, Bi i inne Jednocześnie ważną rolę w kategoryzacji metali ciężkich odgrywają następujące warunki: ich wysoka toksyczność dla organizmów żywych w stosunkowo niskich stężeniach, a także ich zdolność do bioakumulacji i biomagnifikacji. Prawie wszystkie metale objęte tą definicją (z wyjątkiem ołowiu, rtęci, kadmu i bizmutu, których rola biologiczna nie jest obecnie jasna) aktywnie uczestniczą w procesach biologicznych i wchodzą w skład wielu enzymów. Zgodnie z klasyfikacją N. Reimersa metale o gęstości powyżej 8 g/cm3 należy uznać za ciężkie. Tak więc metale ciężkie są Pb, Cu, Zn, Ni, Cd, Co, Sb, Sn, Bi, Hg.
Formalnie zdefiniowane metale ciężkie odpowiada dużej liczbie elementów. Jednak zdaniem badaczy zajmujących się praktycznymi działaniami związanymi z organizacją obserwacji stanu i zanieczyszczenia środowiska, związki tych pierwiastków dalekie są od równorzędnych zanieczyszczeń. Dlatego w wielu pracach następuje zawężenie zakresu grupy metali ciężkich, zgodnie z kryteriami priorytetowymi, ze względu na kierunek i specyfikę pracy. Tak więc w już klasycznych pracach Yu.A. Izrael w wykazie chemikaliów do oznaczenia w podłożach naturalnych na stacjach tła w rezerwatach biosfery, w sekcji metale ciężkie o nazwie Pb, Hg, Cd, As. Z drugiej strony, zgodnie z decyzją Zespołu Zadaniowego ds. Emisji Metali Ciężkich, który działa pod auspicjami Europejskiej Komisji Gospodarczej Organizacji Narodów Zjednoczonych i zbiera oraz analizuje informacje dotyczące emisji zanieczyszczeń w kraje europejskie, tylko Zn, As, Se i Sb zostali przydzieleni do metale ciężkie. Zgodnie z definicją N. Reimersa, obok metali ciężkich wyróżniają się metale szlachetne i rzadkie tylko Pb, Cu, Zn, Ni, Cd, Co, Sb, Sn, Bi, Hg. W pracach stosowanych najczęściej dodawane są metale ciężkie Pt, Ag, W, Fe, Au, Mn.
Jony metali są niezbędnymi składnikami naturalnych zbiorników wodnych. W zależności od warunków środowiskowych (pH, potencjał redoks, obecność ligandów) występują w różnym stopniu utlenienia i wchodzą w skład różnych związków nieorganicznych i metaloorganicznych, które mogą być rzeczywiście rozpuszczone, zdyspergowane koloidalnie lub wchodzić w skład zawiesin mineralnych i organicznych .
Z kolei prawdziwie rozpuszczone formy metali są bardzo zróżnicowane, co wiąże się z procesami hydrolizy, polimeryzacji hydrolitycznej (tworzenie wielopierścieniowych kompleksów hydroksylowych) i kompleksowania różnymi ligandami. W związku z tym zarówno właściwości katalityczne metali, jak i dostępność dla mikroorganizmów wodnych zależą od form ich istnienia w ekosystemie wodnym.
Wiele metali tworzy dość silne kompleksy z materią organiczną; kompleksy te są jedną z najważniejszych form migracji pierwiastków w wodach naturalnych. Większość kompleksów organicznych powstaje w cyklu chelatowym i jest stabilna. Kompleksy utworzone przez kwasy glebowe z solami żelaza, glinu, tytanu, uranu, wanadu, miedzi, molibdenu i innych metali ciężkich są stosunkowo dobrze rozpuszczalne w mediach obojętnych, lekko kwaśnych i lekko zasadowych. Dlatego kompleksy metaloorganiczne mogą migrować w naturalnych wodach na bardzo duże odległości. Jest to szczególnie ważne w przypadku wód niskozmineralizowanych, a przede wszystkim powierzchniowych, w których niemożliwe jest tworzenie się innych kompleksów.
Aby zrozumieć czynniki, które regulują stężenie metali w wodach naturalnych, ich reaktywność chemiczną, biodostępność i toksyczność, konieczne jest poznanie nie tylko całkowitej zawartości, ale także proporcji wolnych i powiązane formularze metal.
Przejście metali w środowisku wodnym do postaci kompleksu metali ma trzy konsekwencje:
1. Może nastąpić wzrost całkowitego stężenia jonów metali z powodu ich przejścia do roztworu z osadów dennych;
2. Przepuszczalność błony jonów złożonych może znacznie różnić się od przepuszczalności jonów uwodnionych;
3. Toksyczność metalu w wyniku kompleksowania może się znacznie zmienić.
Tak więc formy chelatowe Cu, Cd, Hg mniej toksyczny niż wolne jony. Aby zrozumieć czynniki regulujące stężenie metali w wodach naturalnych, ich reaktywność chemiczną, biodostępność i toksyczność, konieczne jest poznanie nie tylko całkowitej zawartości, ale także proporcji form związanych i wolnych.
Źródłami zanieczyszczenia wody metalami ciężkimi są ścieki galwanizernie, przedsiębiorstwa górnicze, hutnictwo żelaza i metali nieżelaznych, zakłady budowy maszyn. Metale ciężkie znajdują się w nawozach i pestycydach i mogą przedostawać się do zbiorników wodnych wraz ze spływaniem z gruntów rolnych.
Wzrost stężenia metali ciężkich w wodach naturalnych często wiąże się z innymi rodzajami zanieczyszczeń, np. zakwaszeniem. Wytrącanie się kwaśnych wytrąceń przyczynia się do obniżenia wartości pH i przejścia metali ze stanu zaadsorbowanego na substancjach mineralnych i organicznych do stanu wolnego.
Przede wszystkim interesujące są te metale, które najbardziej zanieczyszczają atmosferę poprzez ich wykorzystanie w znacznych ilościach w działalności produkcyjnej i w wyniku akumulacji w środowisku zewnętrznym stanowią poważne zagrożenie pod względem ich aktywności biologicznej i właściwości toksycznych . Należą do nich ołów, rtęć, kadm, cynk, bizmut, kobalt, nikiel, miedź, cyna, antymon, wanad, mangan, chrom, molibden i arsen.
Właściwości biogeochemiczne metali ciężkich
H - wysoka, Y - umiarkowana, H - niska
Wanad.
Wanad występuje głównie w stanie rozproszonym i znajduje się w Rudy żelaza, ropa, asfalt, bitum, łupki bitumiczne, węgiel itp. Jednym z głównych źródeł zanieczyszczenia wód naturalnych wanadem jest ropa i produkty jej przetwarzania.
Występuje w wodach naturalnych w bardzo niskich stężeniach: w wodzie rzecznej 0,2 - 4,5 µg/dm3, w wodzie morskiej - średnio 2 µg/dm3
W wodzie tworzy trwałe kompleksy anionowe (V4O12)4- i (V10O26)6-. W migracji wanadu kluczowa jest rola jego rozpuszczonych związków kompleksowych z substancjami organicznymi, zwłaszcza z kwasami huminowymi.
Podwyższone stężenia wanadu są szkodliwe dla zdrowia ludzkiego. MPCv wanadu wynosi 0,1 mg/dm3 (granicznym wskaźnikiem szkodliwości jest sanitarno-toksykologiczny), MPCvr wynosi 0,001 mg/dm3.
Naturalnym źródłem przedostawania się bizmutu do wód naturalnych są procesy ługowania minerałów zawierających bizmut. Źródłem wejścia do wód naturalnych mogą być również ścieki z przemysłu farmaceutycznego i perfumeryjnego, niektóre przedsiębiorstwa przemysłu szklarskiego.
Występuje w niezanieczyszczonych wodach powierzchniowych w stężeniach submikrogramowych. Najwyższe stężenie stwierdzono w wodach gruntowych i wynosi 20 µg/dm3, w wody morskie- 0,02 µg/dm3 MPCv wynosi 0,1 mg/dm3
Głównymi źródłami związków żelaza w wodach powierzchniowych są procesy chemicznego wietrzenia skał, któremu towarzyszy ich mechaniczne niszczenie i rozpuszczanie. W procesie oddziaływania z substancjami mineralnymi i organicznymi zawartymi w wodach naturalnych powstaje złożony kompleks związków żelaza, które znajdują się w wodzie w stanie rozpuszczonym, koloidalnym i zawieszonym. Znaczne ilości żelaza pochodzą ze spływów podziemnych i ścieków z przedsiębiorstw przemysłu metalurgicznego, metalurgicznego, tekstylnego, farbiarskiego i lakierniczego oraz ze ścieków rolniczych.
Równowagi fazowe zależą od składu chemicznego wody, pH, Eh i do pewnego stopnia temperatury. W rutynowej analizie forma ważona emitują cząstki o wielkości powyżej 0,45 mikrona. Są to głównie minerały żelazonośne, hydrat tlenku żelaza i związki żelaza zaadsorbowane w zawiesinach. Prawdziwie rozpuszczona i koloidalna forma jest zwykle rozpatrywana łącznie. Rozpuszczone żelazo reprezentowane przez związki w postaci jonowej, w postaci hydroksokompleksu i kompleksów z rozpuszczonymi substancjami nieorganicznymi i organicznymi wód naturalnych. W formie jonowej migruje głównie Fe(II), a Fe(III) przy braku substancji kompleksujących nie może być w znacznej ilości w stanie rozpuszczonym.
Żelazo znajduje się głównie w wodach o niskich wartościach Eh.
W wyniku chemicznego i biochemicznego (przy udziale bakterii żelazowych) utleniania Fe(II) przechodzi do Fe(III), które po hydrolizie wytrąca się w postaci Fe(OH)3. Zarówno Fe(II), jak i Fe(III) mają tendencję do tworzenia kompleksów hydrokso typu +, 4+, +, 3+, - i inne, które współistnieją w roztworze w różnych stężeniach w zależności od pH i ogólnie określają stan układu żelazo-hydroksyl. Główną formą występowania Fe(III) w wodach powierzchniowych są jego związki kompleksowe z rozpuszczonymi związkami nieorganicznymi i organicznymi, głównie substancjami humusowymi. Przy pH = 8,0 główną formą jest Fe(OH)3 Najsłabiej zbadana jest forma koloidalna żelaza, to uwodniony tlenek żelaza Fe(OH)3 oraz kompleksy z substancjami organicznymi.
Zawartość żelaza w wodach powierzchniowych lądu wynosi dziesiąte części miligrama, w pobliżu bagien - kilka miligramów. Zwiększoną zawartość żelaza obserwuje się w wodach bagiennych, w których występuje w postaci kompleksów z solami kwasów huminowych – humusów. Najwyższe stężenia żelaza (do kilkudziesięciu i setek miligramów na 1 dm3) obserwuje się w wodach gruntowych o niskim pH.
Jako pierwiastek biologicznie czynny, żelazo w pewnym stopniu wpływa na intensywność rozwoju fitoplanktonu oraz skład jakościowy mikroflory w zbiorniku.
Stężenia żelaza podlegają wyraźnym wahaniom sezonowym. Zwykle w zbiornikach o wysokiej produktywności biologicznej, w okresie letniej i zimowej stagnacji, zauważalny jest wzrost stężenia żelaza w dolnych warstwach wody. Jesienno-wiosennemu mieszaniu się mas wody (homotermii) towarzyszy utlenianie Fe(II) do Fe(III) i wytrącanie tego ostatniego w postaci Fe(OH)3.
Wnika do wód naturalnych podczas ługowania gleb, rud polimetalicznych i miedzi, w wyniku rozkładu organizmów wodnych zdolnych do jej akumulacji. Związki kadmu wprowadzane są do wód powierzchniowych ściekami z zakładów ołowiowo-cynkowych, zakładów przeróbczych rud, szeregu przedsiębiorstw chemicznych (produkcja kwasu siarkowego), produkcji galwanicznej, a także z wodami kopalnianymi. Spadek stężenia rozpuszczonych związków kadmu następuje na skutek procesów sorpcji, wytrącania wodorotlenku i węglanu kadmu oraz ich zużycia przez organizmy wodne.
Rozpuszczone formy kadmu w wodach naturalnych to głównie kompleksy mineralne i organiczno-mineralne. Główną zawieszoną formą kadmu są jego adsorbowane związki. Znaczna część kadmu może migrować w komórkach organizmów wodnych.
W wodach rzecznych niezanieczyszczonych i lekko zanieczyszczonych kadm zawiera się w stężeniach submikrogramowych, w wodach zanieczyszczonych i ściekach stężenie kadmu może sięgać kilkudziesięciu mikrogramów w 1 dm3.
Związki kadmu odgrywają ważną rolę w życiu zwierząt i ludzi. Jest toksyczny w wysokich stężeniach, zwłaszcza w połączeniu z innymi substancjami toksycznymi.
MPCv wynosi 0,001 mg/dm3, MPCvr wynosi 0,0005 mg/dm3 (granicą szkodliwości jest toksykologiczna).
Związki kobaltu przedostają się do wód naturalnych w wyniku ich wymywania z pirytu miedzi i innych rud, z gleb podczas rozkładu organizmów i roślin, a także ze ściekami z zakładów hutniczych, hutniczych i chemicznych. Pewne ilości kobaltu pochodzą z gleb w wyniku rozkładu organizmów roślinnych i zwierzęcych.
Związki kobaltu w wodach naturalnych są w stanie rozpuszczonym i zawieszonym, których stosunek ilościowy określa skład chemiczny wody, temperatura i wartości pH. Formy rozpuszczone są reprezentowane głównie przez związki złożone, m.in. z materią organiczną w wodach naturalnych. Najbardziej charakterystyczne dla wód powierzchniowych są związki dwuwartościowego kobaltu. W obecności środków utleniających trójwartościowy kobalt może występować w znacznych stężeniach.
Kobalt jest jednym z biologicznie aktywnych pierwiastków i zawsze znajduje się w ciele zwierząt i roślin. Niewystarczająca zawartość kobaltu w roślinach wiąże się z jego niewystarczającą zawartością w glebach, co przyczynia się do rozwoju anemii u zwierząt (strefa nieczarnoziemno-tajga). Wchodzący w skład witaminy B12 kobalt bardzo aktywnie wpływa na pobór substancji azotowych, wzrost zawartości chlorofilu i kwasu askorbinowego, aktywuje biosyntezę oraz zwiększa zawartość azotu białkowego w roślinach. Jednak podwyższone stężenia związków kobaltu są toksyczne.
W niezanieczyszczonych i lekko zanieczyszczonych wodach rzecznych jego zawartość waha się od dziesiątych do tysięcznych miligrama na 1 dm3, średnia zawartość w wodzie morskiej wynosi 0,5 µg/dm3. MPCv wynosi 0,1 mg/dm3, MPCv wynosi 0,01 mg/dm3.
Mangan
Mangan przedostaje się do wód powierzchniowych w wyniku wymywania rud żelazomanganu i innych minerałów zawierających mangan (piroluzyt, psylomelan, brownit, manganit, czarna ochra). Znaczne ilości manganu pochodzą z rozkładu zwierząt wodnych i organizmów roślinnych, zwłaszcza niebiesko-zielonych, okrzemek i wyższych roślin wodnych. Związki manganu odprowadzane są do zbiorników ze ściekami z zakładów wzbogacania manganu, zakładów metalurgicznych, zakładów przemysłu chemicznego oraz wód kopalnianych.
Spadek stężenia jonów manganu w wodach naturalnych następuje w wyniku utlenienia Mn(II) do MnO2 i innych wytrącających się wysokowartościowych tlenków. Głównymi parametrami determinującymi reakcję utleniania są stężenie rozpuszczonego tlenu, wartość pH i temperatura. Stężenie rozpuszczonych związków manganu zmniejsza się ze względu na ich wykorzystanie przez glony.
Główną formą migracji związków manganu w wodach powierzchniowych są zawiesiny, których skład determinowany jest z kolei składem odwadnianych przez wody skał, a także koloidalnych wodorotlenków metali ciężkich i sorbowanych związków manganu. Istotne znaczenie w migracji manganu w postaci rozpuszczonej i koloidalnej mają substancje organiczne oraz procesy kompleksowego tworzenia manganu z nieorganicznymi i organicznymi ligandami. Mn(II) tworzy rozpuszczalne kompleksy z wodorowęglanami i siarczanami. Kompleksy manganu z jonem chlorkowym są rzadkie. Złożone związki Mn(II) z substancjami organicznymi są zwykle mniej stabilne niż z innymi metalami przejściowymi. Należą do nich związki z aminami, kwasami organicznymi, aminokwasami i substancjami humusowymi. Mn(III) w wysokich stężeniach może być w stanie rozpuszczonym tylko w obecności silnych czynników kompleksujących, Mn(YII) nie występuje w wodach naturalnych.
W wodach rzecznych zawartość manganu waha się zwykle od 1 do 160 µg/dm3, średnia zawartość w wodach morskich to 2 µg/dm3, w wodach podziemnych - 102 - 103 µg/dm3.
Stężenie manganu w wodach powierzchniowych podlega wahaniom sezonowym.
Czynnikami determinującymi zmiany stężeń manganu są stosunek spływu powierzchniowego do podziemnego, intensywność jego zużycia podczas fotosyntezy, rozkład fitoplanktonu, mikroorganizmy i wyższą roślinność wodną, a także procesy jego odkładania się na dnie zbiorników wodnych.
Rola manganu w życiu roślin wyższych i glonów w zbiornikach wodnych jest bardzo duża. Mangan przyczynia się do wykorzystania CO2 przez rośliny, co zwiększa intensywność fotosyntezy, uczestniczy w procesach redukcji azotanów i asymilacji azotu przez rośliny. Mangan wspomaga przejście aktywnego Fe(II) do Fe(III), co chroni komórkę przed zatruciem, przyspiesza wzrost organizmów itp. Ważna ekologiczna i fizjologiczna rola manganu wymaga zbadania i rozmieszczenia manganu w wodach naturalnych.
W przypadku zbiorników wodnych do użytku sanitarnego MPCv (według jonów manganu) ustala się na 0,1 mg/dm3.
Poniżej znajdują się mapy rozkładu średnich stężeń metali: manganu, miedzi, niklu i ołowiu, zbudowane na podstawie danych obserwacyjnych z lat 1989 - 1993. w 123 miastach. Zakłada się, że wykorzystanie nowszych danych jest nieodpowiednie, ponieważ ze względu na zmniejszenie produkcji stężenie zawieszonych ciał stałych, a tym samym metali, znacznie się zmniejszyło.
Wpływ na zdrowie. Wiele metali jest składnikiem pyłu i ma znaczący wpływ na zdrowie.
Mangan dostaje się do atmosfery z emisji z zakładów metalurgii żelaza (60% wszystkich emisji manganu), inżynierii mechanicznej i obróbki metali (23%), metalurgii metali nieżelaznych (9%), wielu małych źródeł, na przykład ze spawania.
Wysokie stężenia manganu prowadzą do pojawienia się efektów neurotoksycznych, postępującego uszkodzenia ośrodkowego system nerwowy, zapalenie płuc.
Najwyższe stężenia manganu (0,57 – 0,66 µg/m3) obserwuje się w dużych ośrodkach hutniczych: w Lipiecku i Czerepowcu oraz w Magadanie. Większość miast o wysokich stężeniach Mn (0,23 - 0,69 µg/m3) koncentruje się na Półwyspie Kolskim: Zapolyarny, Kandalaksha, Monchegorsk, Olenegorsk (patrz mapa).
W latach 1991-1994 emisje manganu ze źródeł przemysłowych zmniejszyły się o 62%, średnie stężenia - o 48%.
Miedź jest jednym z najważniejszych pierwiastków śladowych. Fizjologiczna aktywność miedzi związana jest głównie z włączeniem jej w skład aktywnych centrów enzymów redoks. Niewystarczająca zawartość miedzi w glebie niekorzystnie wpływa na syntezę białek, tłuszczów i witamin oraz przyczynia się do bezpłodności organizmów roślinnych. Miedź bierze udział w procesie fotosyntezy i wpływa na przyswajanie azotu przez rośliny. Jednocześnie nadmierne stężenia miedzi wpływają niekorzystnie na organizmy roślinne i zwierzęce.
W wodach naturalnych najczęściej występują związki Cu(II). Spośród związków Cu(I) najczęściej występują Cu2O, Cu2S i CuCl, które są słabo rozpuszczalne w wodzie. W obecności ligandów w środowisku wodnym, wraz z równowagą dysocjacji wodorotlenków, należy wziąć pod uwagę powstawanie różnych form kompleksowych, które są w równowadze z wodnymi jonami metali.
Głównym źródłem miedzi przedostającej się do wód naturalnych są ścieki z przemysłu chemicznego i metalurgicznego, wody kopalniane oraz odczynniki aldehydowe stosowane do zabijania glonów. Miedź może powstawać w wyniku korozji rur miedzianych i innych konstrukcji stosowanych w instalacjach wodnych. W wodach gruntowych zawartość miedzi wynika z oddziaływania wody ze skałami ją zawierającymi (chalkopiryt, chalkocyt, kowelit, bornit, malachit, azuryt, chryzakola, brotantyna).
Maksymalne dopuszczalne stężenie miedzi w wodzie zbiorników wody sanitarnej i bytowej wynosi 0,1 mg/dm3 (granicznym znakiem szkodliwości jest ogólna sanitarna), w wodzie zbiorników rybackich 0,001 mg/dm3.
Miasto
Norylsk
Monczegorsk
Krasnouralsk
Kołczugino
Zapolyarny
Emisje М (tys. ton/rok) tlenku miedzi i średnie roczne stężenia q (µg/m3) miedzi.
Miedź dostaje się do powietrza z emisjami z przemysłu metalurgicznego. W emisji cząstek stałych występuje głównie w postaci związków, głównie tlenku miedzi.
Przedsiębiorstwa hutnictwa metali nieżelaznych odpowiadają za 98,7% wszystkich antropogenicznych emisji tego metalu, z czego 71% realizują przedsiębiorstwa koncernu Norylsk Nickel zlokalizowane w Zapolyarnym i Nikelu, Monchegorsku i Norylsku, a około 25% emisji miedzi jest realizowanych w Revdzie, Krasnouralsku, Kolchugino i innych.
Wysokie stężenia miedzi prowadzą do zatrucia, anemii i zapalenia wątroby.
Jak widać z mapy, najwyższe stężenia miedzi odnotowuje się w miastach Lipieck i Rudnaja Pristan. Stężenia miedzi wzrosły także w miastach Półwyspu Kolskiego, w Zapolyarnym, Monchegorsku, Nikelu, Olenegorsku, a także w Norylsku.
Emisje miedzi ze źródeł przemysłowych zmniejszyły się o 34%, średnie stężenia - o 42%.
molibden
Związki molibdenu przedostają się do wód powierzchniowych w wyniku ich wymywania z egzogennych minerałów zawierających molibden. Molibden dostaje się również do zbiorników wodnych ze ściekami z zakładów przetwórczych i zakładów metalurgii metali nieżelaznych. Spadek stężeń związków molibdenu następuje w wyniku wytrącania się związków trudno rozpuszczalnych, procesów adsorpcji przez zawiesiny mineralne oraz konsumpcji przez organizmy roślinne.
Molibden w wodach powierzchniowych występuje głównie w postaci MoO42-. Jest wysoce prawdopodobne, że istnieje w postaci kompleksów organiczno-mineralnych. Możliwość pewnej akumulacji w stanie koloidalnym wynika z faktu, że produktami utleniania molibdenitu są sypkie, drobno zdyspergowane substancje.
W wodach rzecznych molibden występuje w stężeniach od 2,1 do 10,6 µg/dm3. Woda morska zawiera średnio 10 µg/dm3 molibdenu.
W niewielkich ilościach molibden jest niezbędny do prawidłowego rozwoju organizmów roślinnych i zwierzęcych. Molibden jest częścią enzymu oksydazy ksantynowej. Przy niedoborze molibdenu enzym powstaje w niewystarczających ilościach, co powoduje negatywne reakcje w organizmie. W wysokich stężeniach molibden jest szkodliwy. Przy nadmiarze molibdenu zaburzony jest metabolizm.
Maksymalne dopuszczalne stężenie molibdenu w zbiornikach wodnych do użytku sanitarnego wynosi 0,25 mg/dm3.
Arszenik dostaje się do wód naturalnych ze źródeł mineralnych, obszarów mineralizacji arsenu (piryty arsenowe, realgar, orpiment), a także ze stref utleniania skał typu polimetalicznego, miedziowo-kobaltowego i wolframowego. Pewna ilość arsenu pochodzi z gleby, a także z rozkładu organizmów roślinnych i zwierzęcych. Spożycie arsenu przez organizmy wodne jest jedną z przyczyn spadku jego stężenia w wodzie, co najwyraźniej objawia się w okresie intensywnego rozwoju planktonu.
Znaczne ilości arsenu przedostają się do zbiorników wodnych ze ściekami z zakładów przetwórczych, odpadów z produkcji barwników, garbarni i fabryk pestycydów, a także z gruntów rolnych, na których stosuje się pestycydy.
W wodach naturalnych związki arsenu są w stanie rozpuszczonym i zawieszonym, których stosunek określa skład chemiczny wody i wartości pH. Arsen w postaci rozpuszczonej występuje w postaci trój- i pięciowartościowej, głównie jako aniony.
W niezanieczyszczonych wodach rzecznych arsen zwykle znajduje się w stężeniach mikrogramowych. W wodach mineralnych jego stężenie może sięgać kilku miligramów na 1 dm3, w wodach morskich zawiera średnio 3 µg/dm3, w wodach podziemnych występuje w stężeniach rzędu 105 µg/dm3. Związki arsenu w wysokich stężeniach są toksyczne dla organizmu zwierząt i ludzi: hamują procesy oksydacyjne, hamują dopływ tlenu do narządów i tkanek.
MPCv dla arsenu wynosi 0,05 mg/dm3 (granicznym wskaźnikiem szkodliwości jest sanitarno-toksykologiczny), a MPCv 0,05 mg/dm3.
Obecność niklu w wodach naturalnych wynika ze składu skał, przez które przepływa woda: znajduje się on w miejscach złóż siarczkowych rud miedziowo-niklowych i rud żelazowo-niklowych. Do wody przedostaje się z gleb oraz z organizmów roślinnych i zwierzęcych podczas ich rozkładu. W sinicach stwierdzono zwiększoną zawartość niklu w porównaniu z innymi rodzajami alg. Związki niklu dostają się również do zbiorników wodnych ze ściekami z zakładów niklowania, zakładów kauczuku syntetycznego i zakładów wzbogacania niklu. Spalaniu paliw kopalnych towarzyszą ogromne emisje niklu.
Jego stężenie może spadać w wyniku wytrącania się związków takich jak cyjanki, siarczki, węglany czy wodorotlenki (przy wzrastających wartościach pH), na skutek jego zużycia przez organizmy wodne oraz procesów adsorpcji.
W wodach powierzchniowych związki niklu występują w stanie rozpuszczonym, zawieszonym i koloidalnym, których stosunek ilościowy zależy od składu wody, temperatury i wartości pH. Sorbentami związków niklu mogą być wodorotlenek żelaza, substancje organiczne, wysoko zdyspergowany węglan wapnia, glinki. Formy rozpuszczone to głównie jony złożone, najczęściej z aminokwasami, kwasami humusowymi i fulwowymi, a także w postaci silnego kompleksu cyjankowego. Związki niklu występują najczęściej w wodach naturalnych, w których znajduje się on na stopniu utlenienia +2. Związki Ni3+ powstają zwykle w środowisku alkalicznym.
Związki niklu odgrywają ważną rolę w procesach krwiotwórczych, będąc katalizatorami. Jego zwiększona zawartość ma specyficzny wpływ na układ krążenia. Nikiel jest jednym z pierwiastków rakotwórczych. Może powodować choroby układu oddechowego. Uważa się, że wolne jony niklu (Ni2+) są około 2 razy bardziej toksyczne niż jego związki kompleksowe.
W niezanieczyszczonych i lekko zanieczyszczonych wodach rzecznych stężenie niklu waha się zwykle od 0,8 do 10 µg/dm3; w zanieczyszczonym jest to kilkadziesiąt mikrogramów na 1 dm3. Średnie stężenie niklu w wodzie morskiej wynosi 2 µg/dm3, w wodach gruntowych – 103 µg/dm3. W wodach podziemnych płuczących skały zawierające nikiel stężenie niklu wzrasta niekiedy do 20 mg/dm3.
Nikiel wchodzi do atmosfery z przedsiębiorstw metalurgii metali nieżelaznych, które odpowiadają za 97% całej emisji niklu, z czego 89% pochodzi z przedsiębiorstw koncernu Norilsk Nickel zlokalizowanych w Zapolyarnym i Nikelu, Monchegorsku i Norylsku.
Podwyższona zawartość niklu w środowisku prowadzi do pojawienia się chorób endemicznych, raka oskrzeli. Związki niklu należą do I grupy czynników rakotwórczych.
Mapa pokazuje kilka punktów o wysokich średnich stężeniach niklu w lokalizacjach koncernu Norylsk Nickel: Apatity, Kandalaksha, Monchegorsk, Olenegorsk.
Emisja niklu z przedsiębiorstw przemysłowych zmniejszyła się o 28%, średnie stężenia - o 35%.
Emisje М (tys. ton/rok) i średnie roczne stężenia q (µg/m3) niklu.
Dostaje się do wód naturalnych w wyniku ługowania minerałów zawierających cynę (kasyteryt, stanina), a także ze ściekami z różnych gałęzi przemysłu (barwienie tkanin, synteza barwników organicznych, produkcja stopów z dodatkiem cyny itp.).
Toksyczny efekt cyny jest niewielki.
Cyna znajduje się w niezanieczyszczonych wodach powierzchniowych w stężeniach submikrogramowych. W wodach gruntowych jego stężenie sięga kilku mikrogramów na 1 dm3. MPCv wynosi 2 mg/dm3.
Związki rtęci mogą przedostawać się do wód powierzchniowych w wyniku wymywania skał w rejonie złóż rtęci (cynober, metacynabaryt, livingstone), w procesie rozkładu organizmów wodnych gromadzących rtęć. Znaczne ilości przedostają się do zbiorników wodnych ze ściekami z przedsiębiorstw produkujących barwniki, pestycydy, farmaceutyki i niektóre materiały wybuchowe. Elektrociepłownie węglowe emitują do atmosfery znaczne ilości związków rtęci, które w wyniku opadów mokrych i suchych przedostają się do zbiorników wodnych.
Spadek stężenia rozpuszczonych związków rtęci następuje w wyniku ich ekstrakcji przez wiele organizmów morskich i słodkowodnych, które mają zdolność akumulowania jej w stężeniach wielokrotnie przewyższających jej zawartość w wodzie, a także procesów adsorpcji przez zawieszone ciała stałe i osady denne.
W wodach powierzchniowych związki rtęci są w stanie rozpuszczonym i zawieszonym. Stosunek między nimi zależy od składu chemicznego wody i wartości pH. Zawieszona rtęć to sorbowane związki rtęci. Formy rozpuszczone to niezdysocjowane cząsteczki, złożone związki organiczne i mineralne. W wodzie zbiorników wodnych rtęć może występować w postaci związków metylortęci.
Związki rtęci są silnie toksyczne, wpływają na układ nerwowy człowieka, powodują zmiany w błonie śluzowej, upośledzenie funkcji motorycznych i wydzielania przewodu pokarmowego, zmiany we krwi itp. Bakteryjne procesy metylacji mają na celu tworzenie związków metylortęci, które są wielokrotnie bardziej toksyczne niż sole mineralne rtęci. Związki metylortęci gromadzą się w rybach i mogą dostać się do organizmu człowieka.
MPCv rtęci wynosi 0,0005 mg/dm3 (granicznym znakiem szkodliwości jest sanitarno-toksykologiczna), MPCv wynosi 0,0001 mg/dm3.
Naturalnymi źródłami ołowiu w wodach powierzchniowych są procesy rozpuszczania minerałów endogennych (galena) i egzogennych (anglesyt, cerusyt itp.). Znaczący wzrost zawartości ołowiu w środowisku (w tym w wodach powierzchniowych) związany jest ze spalaniem węgla, stosowaniem tetraetyloołowiu jako środka przeciwstukowego w paliwie silnikowym, odprowadzaniem do zbiorników wodnych ze ściekami z zakładów przeróbczych rud , niektóre zakłady metalurgiczne, przemysł chemiczny, kopalnie itp. Istotnymi czynnikami wpływającymi na obniżenie stężenia ołowiu w wodzie są jego adsorpcja przez zawiesiny i sedymentacja z nimi do osadów dennych. Wśród innych metali ołów jest wydobywany i akumulowany przez hydrobionty.
Ołów występuje w wodach naturalnych w stanie rozpuszczonym i zawieszonym (sorbowanym). W postaci rozpuszczonej występuje w postaci kompleksów mineralnych i organomineralnych, a także jonów prostych, w postaci nierozpuszczalnej – głównie w postaci siarczków, siarczanów i węglanów.
W wodach rzecznych stężenie ołowiu waha się od dziesiątych części do jednostek mikrogramów na 1 dm3. Nawet w wodach zbiorników przylegających do obszarów rud polimetalicznych jego stężenie rzadko sięga dziesiątek miligramów na 1 dm3. Jedynie w chlorkowych wodach termalnych stężenie ołowiu sięga niekiedy kilku miligramów na 1 dm3.
Granicznym wskaźnikiem szkodliwości ołowiu jest sanitarno-toksykologiczny. MPCv ołowiu wynosi 0,03 mg/dm3, MPCv wynosi 0,1 mg/dm3.
Ołów jest zawarty w emisjach pochodzących z hutnictwa, obróbki metali, elektrotechniki, petrochemii i transportu samochodowego.
Wpływ ołowiu na zdrowie następuje poprzez wdychanie powietrza zawierającego ołów oraz pobranie ołowiu z cząsteczkami pokarmu, wody i kurzu. Ołów gromadzi się w organizmie, w kościach i tkankach powierzchniowych. Ołów wpływa na nerki, wątrobę, układ nerwowy i narządy krwiotwórcze. Osoby starsze i dzieci są szczególnie wrażliwe na nawet małe dawki ołowiu.
Emisje M (tys. ton/rok) i średnie roczne stężenia q (µg/m3) ołowiu.
W ciągu siedmiu lat emisje ołowiu ze źródeł przemysłowych spadły o 60% z powodu cięć produkcji i zamknięcia wielu przedsiębiorstw. Gwałtownemu spadkowi emisji przemysłowych nie towarzyszy spadek emisji z pojazdów. Średnie stężenie ołowiu spadło tylko o 41%. Różnicę we wskaźnikach redukcji emisji i stężeniach ołowiu można wyjaśnić niedoszacowaniem emisji z pojazdów w poprzednich latach; Obecnie wzrosła liczba samochodów i intensywność ich ruchu.
Ołów tetraetylowy
Dostaje się do wód naturalnych ze względu na zastosowanie jako środek przeciwstukowy w paliwie silnikowym pojazdów wodnych, a także ze spływami powierzchniowymi z terenów zurbanizowanych.
Substancja ta charakteryzuje się wysoką toksycznością, ma właściwości kumulacyjne.
Źródłem srebra dostającego się do wód powierzchniowych są wody gruntowe i ścieki z kopalń, zakładów przetwórczych i zakładów fotograficznych. Podwyższona zawartość srebra związana jest ze stosowaniem preparatów bakteriobójczych i glonobójczych.
W ściekach srebro może występować w postaci rozpuszczonej i zawieszonej, głównie w postaci soli halogenkowych.
W niezanieczyszczonych wodach powierzchniowych srebro występuje w stężeniach submikrogramowych. W wodach gruntowych stężenie srebra waha się od kilku do kilkudziesięciu mikrogramów w 1 dm3, w wodzie morskiej średnio 0,3 μg/dm3.
Jony srebra potrafią niszczyć bakterie i sterylizować wodę nawet w niewielkich stężeniach (dolna granica bakteriobójczego działania jonów srebra to 2,10-11 mol/dm3). Rola srebra w organizmie zwierząt i ludzi nie została dostatecznie zbadana.
MPCv srebra wynosi 0,05 mg/dm3.
Antymon przedostaje się do wód powierzchniowych poprzez ługowanie minerałów antymonu (stibnit, senarmontit, walentyny, serynit, stibiokanit) oraz ze ściekami z zakładów gumowych, szklarskich, farbiarskich i zapałek.
W wodach naturalnych związki antymonu są w stanie rozpuszczonym i zawieszonym. W warunkach redoks charakterystycznych dla wód powierzchniowych może występować zarówno trójwartościowy, jak i pięciowartościowy antymon.
W wodach powierzchniowych niezanieczyszczonych antymon występuje w stężeniach submikrogramowych, w wodzie morskiej jego stężenie sięga 0,5 µg/dm3, w wodach podziemnych 10 µg/dm3. MPCv antymonu wynosi 0,05 mg/dm3 (granicznym wskaźnikiem szkodliwości jest sanitarno-toksykologiczny), MPCv wynosi 0,01 mg/dm3.
Trój- i sześciowartościowe związki chromu przedostają się do wód powierzchniowych w wyniku wymywania ze skał (chromit, krokoit, uwarowit itp.). Pewne ilości pochodzą z rozkładu organizmów i roślin, z gleb. Znaczne ilości mogą przedostawać się do zbiorników wodnych ze ściekami z galwanizerni, farbiarni przedsiębiorstw włókienniczych, garbarni i przemysłu chemicznego. Spadek stężenia jonów chromu można zaobserwować w wyniku ich zużycia przez organizmy wodne oraz procesów adsorpcji.
W wodach powierzchniowych związki chromu są w stanie rozpuszczonym i zawieszonym, których stosunek zależy od składu wody, temperatury i pH roztworu. Zawieszone związki chromu to głównie sorbowane związki chromu. Sorbentami mogą być glinki, wodorotlenek żelaza, silnie zdyspergowany osadzający się węglan wapnia, pozostałości roślinne i zwierzęce. W postaci rozpuszczonej chrom może występować w postaci chromianów i dichromianów. W warunkach tlenowych Cr(VI) przekształca się w Cr(III), którego sole w środowisku obojętnym i zasadowym ulegają hydrolizie z uwolnieniem wodorotlenku.
W niezanieczyszczonych i lekko zanieczyszczonych wodach rzecznych zawartość chromu waha się od kilku dziesiątych mikrograma na litr do kilku mikrogramów na litr, w zanieczyszczonych zbiornikach wodnych sięga kilkudziesięciu i setek mikrogramów na litr. Średnie stężenie w wodach morskich wynosi 0,05 µg/dm3, w wodach gruntowych - zwykle w granicach 10 - 102 µg/dm3.
Związki Cr(VI) i Cr(III) w zwiększonych ilościach wykazują właściwości rakotwórcze. Bardziej niebezpieczne są związki Cr(VI).
Dostaje się do wód naturalnych w wyniku naturalnych procesów niszczenia i rozpuszczania skał i minerałów (sfaleryt, cynkit, goslaryt, smithsonit, kalamina), a także ze ściekami z zakładów przeróbki rudy i galwanizerni, produkcji pergaminu, farb mineralnych , włókno wiskozowe i inne
W wodzie występuje głównie w postaci jonowej lub w postaci jej kompleksów mineralno-organicznych. Czasami występuje w formach nierozpuszczalnych: w postaci wodorotlenku, węglanu, siarczku itp.
W wodach rzecznych stężenie cynku wynosi zwykle od 3 do 120 µg/dm3, w wodach morskich od 1,5 do 10 µg/dm3. Zawartość w rudzie, a zwłaszcza w wodach kopalnianych o niskim pH, może być znaczna.
Cynk jest jednym z aktywnych pierwiastków śladowych, które wpływają na wzrost i prawidłowy rozwój organizmów. Jednocześnie wiele związków cynku jest toksycznych, przede wszystkim jego siarczan i chlorek.
MPCv Zn2+ wynosi 1 mg/dm3 (wskaźnik graniczny szkodliwości - organoleptyczny), MPCvr Zn2+ - 0,01 mg/dm3 (objaw graniczny szkodliwości - toksykologiczny).
Metale ciężkie są już na drugim miejscu pod względem zagrożenia, ustępując pestycydom i znacznie wyprzedzając tak znane zanieczyszczenia, jak dwutlenek węgla i siarka, ale w prognozie powinny stać się najniebezpieczniejsze, groźniejsze niż odpady i ciała stałe z elektrowni jądrowych. marnotrawstwo. Zanieczyszczenie metalami ciężkimi wiąże się z ich powszechnym stosowaniem w produkcji przemysłowej, połączonym ze słabymi systemami oczyszczania, w wyniku czego metale ciężkie przedostają się do środowiska, w tym do gleby, zanieczyszczając je i zatruwając.
Metale ciężkie należą do priorytetowych zanieczyszczeń, których monitorowanie jest obowiązkowe we wszystkich środowiskach. W różnych pracach naukowych i stosowanych autorzy w różny sposób interpretują znaczenie pojęcia „metali ciężkich”. W niektórych przypadkach definicja metali ciężkich obejmuje pierwiastki, które są kruche (na przykład bizmut) lub metaloidy (na przykład arsen).
Gleba jest głównym medium, do którego przedostają się metale ciężkie, w tym z atmosfery i środowiska wodnego. Służy również jako źródło wtórnego zanieczyszczenia powietrza powierzchniowego i wód, które dostają się z niego do Oceanu Światowego. Metale ciężkie są asymilowane z gleby przez rośliny, które następnie dostają się do pokarmu lepiej zorganizowanych zwierząt.
kontynuacja
--PODZIAŁ STRONY-- 3.3. zatrucie ołowiem
Obecnie ołów zajmuje pierwsze miejsce wśród przyczyn zatruć przemysłowych. Wynika to z szerokiego zastosowania w różnych gałęziach przemysłu. Pracownicy rudy ołowiu są narażeni na kontakt z ołowiem w hutach ołowiu, przy produkcji baterii, przy lutowaniu, w drukarniach, przy produkcji szkła kryształowego lub wyrobów ceramicznych, benzyny ołowiowej, farb ołowiowych itp. Zanieczyszczenie ołowiem powietrze atmosferyczne, gleba i woda w sąsiedztwie takich przemysłów, a także w pobliżu głównych autostrad, stwarza zagrożenie narażenia na ołów ludności zamieszkującej te tereny, a przede wszystkim dzieci, które są bardziej wrażliwe na działanie metali ciężkich.
Z przykrością należy zauważyć, że w Rosji nie ma polityki państwa w zakresie prawnej, regulacyjnej i ekonomicznej regulacji wpływu ołowiu na środowisko i zdrowie publiczne, ograniczania emisji (zrzutów, odpadów) ołowiu i jego związków do środowiska , oraz o całkowitym zaprzestaniu produkcji benzyny zawierającej ołów.
Ze względu na skrajnie niesatysfakcjonującą pracę edukacyjną wyjaśniającą społeczeństwu stopień niebezpieczeństwa narażenia organizmu ludzkiego na metale ciężkie, w Rosji liczba kontyngentów mających zawodowy kontakt z ołowiem nie maleje, ale stopniowo rośnie. Przypadki chronicznego zatrucia ołowiem zostały zarejestrowane w 14 branżach w Rosji. Wiodące branże to przemysł elektryczny (produkcja baterii), oprzyrządowanie, drukowanie i metalurgia metali nieżelaznych, w nich zatrucie jest spowodowane przekroczeniem 20 lub więcej razy maksymalnego dopuszczalnego stężenia (MPC) ołowiu w powietrzu obszaru roboczego.
Istotnym źródłem ołowiu są spaliny samochodowe, ponieważ połowa Rosji nadal używa benzyny ołowiowej. Głównym źródłem zanieczyszczenia środowiska pozostają jednak zakłady metalurgiczne, w szczególności huty miedzi. A tu są liderzy. Na terytorium Obwód swierdłowski w kraju istnieją 3 największe źródła emisji ołowiu: w miastach Krasnouralsk, Kirovograd i Revda.
Kominy huty miedzi w Krasnouralsku, zbudowanej w latach stalinowskiej industrializacji i wykorzystującej sprzęt z 1932 r., wyrzucają rocznie 150-170 ton ołowiu do 34-tysięcznego miasta, pokrywając wszystko ołowianym pyłem.
Stężenie ołowiu w glebie Krasnouralska waha się od 42,9 do 790,8 mg/kg przy maksymalnym dopuszczalnym stężeniu MPC = 130 mikronów/kg. Próbki wody w wodociągu sąsiedniej wsi. Oktiabrsky, zasilany z podziemnego źródła wody, odnotował nawet dwukrotny nadmiar RPP.
Zanieczyszczenie ołowiem ma wpływ na zdrowie człowieka. Narażenie na ołów zaburza żeński i męski układ rozrodczy. Dla kobiet w ciąży i w wieku rozrodczym szczególnie niebezpieczny jest podwyższony poziom ołowiu we krwi, ponieważ ołów zaburza czynności menstruacyjne, częściej dochodzi do przedwczesnych porodów, poronień i śmierci płodów z powodu przenikania ołowiu przez barierę łożyskową. Noworodki mają wysoką śmiertelność.
Zatrucie ołowiem jest niezwykle niebezpieczne dla małych dzieci – wpływa na rozwój mózgu i układu nerwowego. Badanie 165 dzieci w Krasnouralsku w wieku od 4 lat wykazało znaczne upośledzenie umysłowe u 75,7%, au 6,8% przebadanych dzieci stwierdzono upośledzenie umysłowe, w tym upośledzenie umysłowe.
Dzieci wiek przedszkolny są najbardziej podatne na szkodliwe działanie ołowiu, ponieważ ich układ nerwowy jest w trakcie tworzenia. Nawet w małych dawkach zatrucie ołowiem powoduje spadek rozwoju intelektualnego, uwagi i zdolności koncentracji, opóźnienie w czytaniu, prowadzi do rozwoju agresywności, nadpobudliwości i innych problemów behawioralnych u dziecka. Te nieprawidłowości rozwojowe mogą być długotrwałe i nieodwracalne. Niska masa urodzeniowa, zahamowanie wzrostu i utrata słuchu są również wynikiem zatrucia ołowiem. Wysokie dawki zatrucia prowadzą do upośledzenia umysłowego, śpiączki, drgawek i śmierci.
Biała księga opublikowana przez rosyjskich ekspertów donosi, że zanieczyszczenie ołowiem obejmuje cały kraj i jest jedną z wielu katastrof ekologicznych w byłym Związku Radzieckim, które wyszły na jaw w ostatnich latach. Większość terytorium Rosji jest obciążona opadem ołowiu, która przekracza wartość krytyczną dla normalnego funkcjonowania ekosystemu. W kilkudziesięciu miastach występuje nadmiar stężeń ołowiu w powietrzu i glebie powyżej wartości odpowiadających MPC.
Najwyższy poziom zanieczyszczenia powietrza ołowiem, przekraczający RPP, zaobserwowano w miastach Komsomolsk nad Amurem, Tobolsk, Tiumeń, Karabash, Władimir, Władywostok.
Maksymalne ładunki depozycji ołowiu prowadzące do degradacji ekosystemów lądowych obserwowane są w rejonach Moskwy, Włodzimierza, Niżnego Nowogrodu, Riazania, Tuły, Rostowa i Leningradu.
Źródła stacjonarne odpowiadają za zrzut ponad 50 ton ołowiu w postaci różnych związków do zbiorników wodnych. Jednocześnie 7 fabryk akumulatorów zrzuca rocznie przez system kanalizacyjny 35 ton ołowiu. Analiza rozmieszczenia zrzutów ołowiu do zbiorników wodnych na terytorium Rosji pokazuje, że liderami tego typu ładunku są regiony Leningradu, Jarosławia, Permu, Samary, Penzy i Oryola.
Kraj potrzebuje pilnych środków w celu zmniejszenia zanieczyszczenia ołowiem, ale jak dotąd kryzys gospodarczy w Rosji przyćmiewa problemy środowiskowe. W przedłużającym się kryzysie przemysłowym Rosji brakuje funduszy na usuwanie zanieczyszczeń z przeszłości, ale jeśli gospodarka zacznie się poprawiać, a fabryki wrócą do pracy, zanieczyszczenie może się tylko pogorszyć.
10 najbardziej zanieczyszczonych miast byłego ZSRR
(Metale są wymienione w porządku malejącym według poziomu priorytetu dla danego miasta)
4. Higiena gleby. Utylizacja odpadów.
Gleba w miastach i innych osadach oraz ich okolicach od dawna różni się od naturalnej, biologicznie cennej gleby, która odgrywa ważną rolę w utrzymaniu równowagi ekologicznej. Gleba w miastach podlega takim samym szkodliwym wpływom jak powietrze miejskie i hydrosfera, dlatego wszędzie następuje jej znaczna degradacja. Higienie gleby nie poświęca się wystarczającej uwagi, chociaż jej znaczenie jako jednego z głównych składników biosfery (powietrza, wody, gleby) i biologicznego czynnika środowiskowego jest nawet ważniejsze niż wody, gdyż jej ilość (przede wszystkim jakość wody gruntowe) zależy od stanu gleby i nie można oddzielić tych czynników od siebie. Gleba ma zdolność biologicznego samooczyszczania: w glebie następuje rozszczepienie wpadających do niej odpadów i ich mineralizacja; w końcu gleba rekompensuje utracone minerały ich kosztem.
Jeżeli w wyniku przeciążenia gleby którykolwiek ze składników jej zdolności mineralizacyjnej zostanie utracony, nieuchronnie doprowadzi to do naruszenia mechanizmu samooczyszczania i całkowitej degradacji gleby. Przeciwnie, stworzenie optymalnych warunków do samooczyszczania gleby przyczynia się do zachowania równowagi ekologicznej i warunków istnienia wszystkich żywych organizmów, w tym ludzi.
Dlatego problem unieszkodliwiania odpadów o szkodliwym działaniu biologicznym nie ogranicza się do kwestii ich eksportu; jest to bardziej złożony problem higieniczny, ponieważ gleba jest łącznikiem między wodą, powietrzem i człowiekiem.
4.1.
Rola gleby w metabolizmie
Biologiczny związek między glebą a człowiekiem odbywa się głównie poprzez metabolizm. Gleba jest niejako dostawcą minerałów niezbędnych do cyklu metabolicznego, do wzrostu roślin spożywanych przez ludzi i roślinożerców, zjadanych kolejno przez ludzi i mięsożerców. W ten sposób gleba dostarcza pożywienia wielu przedstawicielom świata roślinnego i zwierzęcego.
W konsekwencji pogorszenie jakości gleby, spadek jej wartości biologicznej, jej zdolności do samooczyszczania powoduje reakcję łańcuchową, która w przypadku długotrwałych szkodliwych skutków może prowadzić do różnych zaburzeń zdrowotnych wśród populacji. Ponadto w przypadku spowolnienia procesów mineralizacji azotany, azot, fosfor, potas itp. powstające podczas rozpadu substancji mogą przedostać się do wód gruntowych używanych do picia i powodować poważne choroby (np. azotany mogą powodować methemoglobinemię, głównie u niemowląt). .
Spożywanie wody z gleby ubogiej w jod może powodować endemiczne wole itp.
4.2.
Ekologiczny związek między glebą a wodą i odpadami płynnymi (ściekami)
Człowiek wydobywa z gleby wodę niezbędną do utrzymania procesów metabolicznych i samego życia. Jakość wody zależy od stanu gleby; zawsze odzwierciedla stan biologiczny danej gleby.
Dotyczy to w szczególności wód podziemnych, których wartość biologiczna jest zasadniczo determinowana właściwościami gleby i gleby, zdolnością tych ostatnich do samooczyszczania się, zdolnością filtracyjną, składem makroflory, mikrofauną itp.
Bezpośredni wpływ gleby na wody powierzchniowe jest już mniej znaczący, związany jest głównie z opadami atmosferycznymi. Na przykład po ulewnych deszczach różne zanieczyszczenia są wypłukiwane z gleby do otwartych zbiorników wodnych (rzeki, jeziora), w tym sztuczne nawozy (azot, fosforany), pestycydy, herbicydy; w obszarach krasowych, spękanych osadów mogą przenikać zanieczyszczenia pęka w głąb wód gruntowych.
Nieodpowiednie oczyszczanie ścieków może również powodować szkodliwe skutki biologiczne dla gleby i ostatecznie prowadzić do degradacji gleby. Dlatego ochrona gleby w osiedlach jest jednym z głównych wymogów ochrony środowiska w ogóle.
4.3.
Limity obciążenia gleby odpadami stałymi (odpady z gospodarstw domowych i ulic, odpady przemysłowe, suche osady z sedymentacji ścieków, substancje promieniotwórcze itp.)
Problem pogłębia fakt, że w wyniku wytwarzania w miastach coraz większej ilości odpadów stałych gleba w ich sąsiedztwie poddawana jest coraz większej presji. W coraz szybszym tempie pogarszają się właściwości i skład gleby.
Z 64,3 mln ton papieru wyprodukowanego w USA 49,1 mln ton trafia do odpadów (z tej ilości 26 mln ton dostarcza gospodarstwo domowe, a 23,1 mln ton sieć handlowa).
W związku z powyższym wywóz i ostateczne unieszkodliwianie odpadów stałych jest bardzo istotnym, trudniejszym do realizacji problemem higienicznym w kontekście postępującej urbanizacji.
Możliwa jest ostateczna utylizacja odpadów stałych w zanieczyszczonej glebie. Jednak ze względu na stale pogarszającą się zdolność samooczyszczania się gleby miejskiej, ostateczna utylizacja zakopanych w ziemi odpadów jest niemożliwa.
Człowiek mógłby z powodzeniem wykorzystać procesy biochemiczne zachodzące w glebie, jej neutralizującą i dezynfekującą zdolność do unieszkodliwiania odpadów stałych, jednak gleba miejska, w wyniku wielowiekowego zamieszkiwania przez człowieka w miastach i jego działalności, już dawno nie nadaje się do tego celu.
Dobrze znane są mechanizmy samooczyszczania, mineralizacji zachodzącej w glebie, rola bakterii i enzymów w nich zaangażowanych oraz produktów pośrednich i końcowych rozkładu substancji. Obecnie badania mają na celu identyfikację czynników zapewniających równowagę biologiczną naturalnej gleby, a także wyjaśnienie, ile odpadów stałych (i jaki skład) może prowadzić do naruszenia równowagi biologicznej gleby.
Ilość odpadów domowych (śmieci) na mieszkańca niektórych dużych miast świata
Należy zauważyć, że stan higieniczny gleby w miastach w wyniku jej przeciążenia gwałtownie się pogarsza, chociaż zdolność gleby do samooczyszczania jest głównym wymogiem higienicznym dla zachowania równowagi biologicznej. Gleba w miastach nie jest już w stanie poradzić sobie ze swoim zadaniem bez pomocy człowieka. Jedyne wyjście z stworzonego przepisy - pełne unieszkodliwianie i niszczenie odpadów zgodnie z wymogami higienicznymi.
Dlatego budowa obiektów użyteczności publicznej powinna mieć na celu zachowanie naturalnej zdolności gleby do samooczyszczania, a jeśli ta zdolność już stała się niezadowalająca, należy ją sztucznie przywrócić.
Najbardziej niekorzystne jest toksyczne działanie odpadów przemysłowych, zarówno płynnych, jak i stałych. Do gleby trafia coraz więcej takich odpadów, z którymi nie jest w stanie sobie poradzić. I tak np. zanieczyszczenie gleby arsenem stwierdzono w sąsiedztwie zakładów produkujących superfosfaty (w promieniu 3 km). Jak wiadomo, niektóre pestycydy, takie jak związki chloroorganiczne, które dostały się do gleby, nie rozkładają się przez długi czas.
Podobnie sytuacja wygląda w przypadku niektórych syntetycznych materiałów opakowaniowych (polichlorek winylu, polietylen itp.).
Niektóre toksyczne związki prędzej czy później dostają się do wód gruntowych, w wyniku czego nie tylko zostaje zaburzona biologiczna równowaga gleby, ale także jakość wód gruntowych pogarsza się do tego stopnia, że nie można ich już używać jako wody pitnej.
Procent liczby głównych materiały syntetyczne zawarte w odpadach domowych (śmieci)
*
Razem z odpadami innych tworzyw sztucznych, które twardnieją pod wpływem ciepła.
Problem odpadów nasilił się dziś również dlatego, że część odpadów, głównie odchody ludzi i zwierząt, jest wykorzystywana do nawożenia gruntów rolnych [odchody zawierają znaczną ilość azotu – 0,4-0,5%, fosforu (P203) – 0,2-0,6 %, potas (K>0) -0,5-1,5%, węgiel-5-15%]. Ten problem miasta rozprzestrzenił się na dzielnice miasta.
4.4.
Rola gleby w rozprzestrzenianiu się różnych chorób
Gleba odgrywa rolę w rozprzestrzenianiu się chorób zakaźnych. Poinformowali o tym w ubiegłym stuleciu Petterkoffer (1882) i Fodor (1875), którzy podkreślali głównie rolę gleby w rozprzestrzenianiu się chorób jelitowych: cholery, tyfusu, czerwonki itp. Zwrócili również uwagę na fakt, że niektórzy bakterie i wirusy pozostają żywotne i zjadliwe w glebie przez wiele miesięcy. Następnie wielu autorów potwierdziło swoje obserwacje, zwłaszcza w odniesieniu do gleby miejskiej. Na przykład czynnik sprawczy cholery pozostaje żywy i patogenny w wodach gruntowych od 20 do 200 dni, czynnik sprawczy tyfusu w kale - od 30 do 100 dni, czynnik sprawczy paratyfusu - od 30 do 60 dni. (Pod względem rozprzestrzeniania się chorób zakaźnych gleba miejska jest znacznie bardziej niebezpieczna niż gleba polna nawożona obornikiem.)
Do określenia stopnia zanieczyszczenia gleby wielu autorów stosuje oznaczanie liczby bakterii (E. coli), podobnie jak przy określaniu jakości wody. Inni autorzy uznają za celowe określenie dodatkowo liczby bakterii termofilnych biorących udział w procesie mineralizacji.
Rozprzestrzenianie się chorób zakaźnych przez glebę jest znacznie ułatwione dzięki nawadnianiu gleby ściekami. Jednocześnie pogarszają się również właściwości mineralizacyjne gleby. Dlatego podlewanie ściekami powinno odbywać się pod stałym ścisłym nadzorem sanitarnym i tylko poza obszarem miejskim.
4.5.
Szkodliwe działanie głównych rodzajów zanieczyszczeń (odpadów stałych i płynnych) prowadzących do degradacji gleby
4.5.1.
Neutralizacja odpadów płynnych w glebie
W liczbie rozliczenia bez kanalizacji niektóre odpady, w tym obornik, są unieszkodliwiane w glebie.
Jak wiesz, to najłatwiejszy sposób na zneutralizowanie. Jest to jednak dopuszczalne tylko wtedy, gdy mamy do czynienia z glebą biologicznie cenną, która zachowała zdolność do samooczyszczania, co nie jest typowe dla gleb miejskich. Jeżeli gleba nie posiada już tych właściwości, to aby chronić ją przed dalszą degradacją, potrzebne jest kompleksowe zaplecze techniczne do unieszkodliwiania płynnych ścieków.
W wielu miejscach odpady unieszkodliwiane są w dołach kompostowych. Technicznie to rozwiązanie jest trudne zadanie. Ponadto płyny są w stanie wnikać w glebę na dość duże odległości. Zadanie komplikuje dodatkowo fakt, że ścieki miejskie zawierają coraz większą ilość toksycznych odpadów przemysłowych, które pogarszają właściwości mineralizacyjne gleby w jeszcze większym stopniu niż odchody ludzi i zwierząt. Dlatego w doły kompostowe dopuszczalne jest odprowadzanie tylko ścieków, które zostały wstępnie osadzone. W przeciwnym razie zdolność filtracyjna gleby jest zaburzona, wówczas gleba traci inne właściwości ochronne, pory stopniowo się blokują itp.
Wykorzystanie ludzkich odchodów do nawadniania pól uprawnych jest drugim sposobem unieszkodliwiania płynnych odpadów. Ta metoda stwarza podwójne zagrożenie higieniczne: po pierwsze może prowadzić do przeciążenia gleby, po drugie odpady te mogą stać się poważnym źródłem infekcji. Dlatego kał musi być najpierw zdezynfekowany i poddany odpowiedniej obróbce, a dopiero potem wykorzystany jako nawóz. Są tu dwa przeciwstawne punkty widzenia. Zgodnie z wymogami higienicznymi odchody ulegają niemal całkowitemu zniszczeniu iz punktu widzenia gospodarki narodowej stanowią cenny nawóz. Świeże odchody nie mogą być używane do podlewania ogrodów i pól bez uprzedniej ich dezynfekcji. Jeśli nadal musisz używać świeżych odchodów, wymagają one takiego stopnia neutralizacji, że prawie nie mają wartości jako nawóz.
Odchody mogą być stosowane jako nawóz tylko w specjalnie wyznaczonych miejscach - przy stałej kontroli sanitarno-higienicznej, zwłaszcza pod kątem stanu wód gruntowych, ilości much itp.
Wymagania dotyczące usuwania i usuwania odchodów zwierzęcych do gleby nie różnią się zasadniczo od wymagań dotyczących usuwania odchodów ludzkich.
Do niedawna obornik był znaczącym źródłem cennych składników odżywczych dla rolnictwa, poprawiających żyzność gleby. Jednak w ostatnich latach obornik stracił na znaczeniu częściowo z powodu mechanizacji rolnictwa, a częściowo z powodu rosnącego stosowania nawozów sztucznych.
W przypadku braku odpowiedniej obróbki i utylizacji niebezpieczny jest również obornik, a także nieoczyszczony ludzki kał. Dlatego przed zabraniem na pola obornik może dojrzewać, aby w tym czasie (w temperaturze 60-70 ° C) mogły w nim zachodzić niezbędne procesy biotermiczne. Następnie obornik jest uważany za „dojrzały” i wolny od większości zawartych w nim patogenów (bakterie, jaja robaków itp.).
Należy pamiętać, że magazyny obornika mogą stanowić idealne pożywki dla much, które sprzyjają rozprzestrzenianiu się różnych infekcji jelitowych. Należy zauważyć, że muchy do reprodukcji najchętniej wybierają obornik świński, następnie koński, owczy i wreszcie krowi. Przed wywiezieniem obornika na pola należy go poddać działaniu środków owadobójczych.
kontynuacja
--PODZIAŁ STRONY--
Metale ciężkie w glebie
W ostatnim czasie, w związku z szybkim rozwojem przemysłu, nastąpił znaczny wzrost poziomu metali ciężkich w środowisku. Termin „metale ciężkie” stosuje się do metali o gęstości przekraczającej 5 g/cm3 lub o liczbie atomowej większej niż 20. Chociaż istnieje inny punkt widzenia, zgodnie z którym ponad 40 pierwiastki chemiczne o masach atomowych większych niż 50 at. jednostki Wśród pierwiastków chemicznych metale ciężkie są najbardziej toksyczne i pod względem poziomu zagrożenia ustępują im jedynie pestycydów. Jednocześnie toksyczne są następujące pierwiastki chemiczne: Co, Ni, Cu, Zn, Sn, As, Se, Te, Rb, Ag, Cd, Au, Hg, Pb, Sb, Bi, Pt.
Fitotoksyczność metali ciężkich zależy od ich właściwości chemiczne: wartościowość, promień jonowy i zdolność do tworzenia kompleksów. W większości przypadków, w zależności od stopnia toksyczności, pierwiastki ułożone są w kolejności: Cu> Ni> Cd> Zn> Pb> Hg> Fe> Mo> Mn. Jednak seria ta może się nieco zmienić ze względu na nierównomierne wytrącanie się pierwiastków przez glebę i przejście w stan niedostępny dla roślin, warunki wzrostu oraz cechy fizjologiczne i genetyczne samych roślin. Przemiana i migracja metali ciężkich następuje pod bezpośrednim i pośrednim wpływem reakcji tworzenia kompleksu. Przy ocenie zanieczyszczenia środowiska należy wziąć pod uwagę właściwości gleby, a przede wszystkim skład granulometryczny, zawartość próchnicy i buforowanie. Przez zdolność buforową rozumie się zdolność gleb do utrzymywania stężenia metali w roztworze glebowym na stałym poziomie.
W glebach metale ciężkie występują w dwóch fazach – stałej iw roztworze glebowym. O formie istnienia metali decyduje odczyn środowiska, skład chemiczny i materiałowy roztworu glebowego, a przede wszystkim zawartość substancji organicznych. Pierwiastki - kompleksanty zanieczyszczające glebę skoncentrowane są głównie w jej górnej warstwie 10 cm. Jednak w przypadku zakwaszenia gleby niskobuforowej znaczna część metali ze stanu pochłaniania wymiennego przechodzi do roztworu glebowego. Kadm, miedź, nikiel, kobalt mają silną zdolność migracji w środowisku kwaśnym. Spadek pH o 1,8-2 jednostki prowadzi do zwiększenia ruchliwości cynku o 3,8-5,4, kadmu - o 4-8, miedzi - o 2-3 razy.
Tabela 1 Standardy MPC (MAC), stężenia tła pierwiastków chemicznych w glebie (mg/kg)
| Element | Klasa zagrożenia | RPP | AEC według grup gleb | treść w tle | |||
| Zawartość brutto | Ekstrahowalny buforem octanu amonu (pН=4,8) | Piaszczysta, piaszczysta | gliniasty, gliniasty | ||||
| pH ks l< 5,5 | pH ks l > 5,5 | ||||||
| Pb | 1 | 32 | 6 | 32 | 65 | 130 | 26 |
| Zn | 1 | - | 23 | 55 | 110 | 220 | 50 |
| płyta CD | 1 | - | - | 0,5 | 1 | 2 | 0,3 |
| Cu | 2 | - | 3 | 33 | 66 | 132 | 27 |
| Ni | 2 | - | 4 | 20 | 40 | 80 | 20 |
| Więc | 2 | - | 5 | - | - | - | 7,2 |
W związku z tym metale ciężkie po wejściu do gleby szybko wchodzą w interakcję z ligandami organicznymi, tworząc złożone związki. Tak więc przy niskich stężeniach w glebie (20-30 mg/kg) około 30% ołowiu występuje w postaci kompleksów z substancjami organicznymi. Udział związków kompleksowych ołowiu wzrasta wraz ze stężeniem do 400 mg/g, a następnie maleje. Metale są również sorbowane (wymiana lub nie) przez wytrącanie wodorotlenków żelaza i manganu, minerałów ilastych oraz materii organicznej gleby. Metale dostępne dla roślin i zdolne do ługowania znajdują się w roztworze glebowym w postaci wolnych jonów, kompleksów i chelatów.
Pobieranie HMs przez glebę w większym stopniu zależy od reakcji środowiska i tego, jakie aniony przeważają w roztworze glebowym. W środowisku kwaśnym miedź, ołów i cynk są bardziej sorbowane, a w alkalicznym kadm i kobalt są intensywnie wchłaniane. Miedź preferencyjnie wiąże się z ligandami organicznymi i wodorotlenkami żelaza.
Tabela 2 Mobilność pierwiastków śladowych w różnych glebach w zależności od pH roztworu glebowego
Czynniki glebowo-klimatyczne często determinują kierunek i tempo migracji i przemian HMs w glebie. Tak więc warunki glebowo-wodne strefy leśno-stepowej przyczyniają się do intensywnej pionowej migracji HM wzdłuż profilu glebowego, w tym możliwego przenoszenia metali z przepływem wody wzdłuż pęknięć, cieków korzeniowych itp.
Nikiel (Ni) jest pierwiastkiem grupy VIII układu okresowego o masie atomowej 58,71. Nikiel wraz z Mn, Fe, Co i Cu należy do tzw. metali przejściowych, których związki są wysoce aktywne biologicznie. Ze względu na specyfikę budowy orbitali elektronowych powyższe metale, w tym nikiel, mają wyraźnie zaznaczoną zdolność do tworzenia kompleksów. Nikiel jest w stanie tworzyć stabilne kompleksy np. z cysteiną i cytrynianem, a także z wieloma ligandami organicznymi i nieorganicznymi. Skład geochemiczny skał macierzystych w dużej mierze determinuje zawartość niklu w glebach. Najwięcej niklu znajduje się w glebach wytworzonych ze skał podstawowych i ultrazasadowych. Według niektórych autorów limity nadmiernego i toksycznego poziomu niklu dla większości gatunków wahają się od 10 do 100 mg/kg. Główna masa niklu jest nieruchomo osadzona w glebie, a bardzo słaba migracja w stanie koloidalnym oraz w składzie zawiesin mechanicznych nie wpływa na ich rozkład wzdłuż profilu pionowego i jest dość równomierna.
Ołów (Pb). Chemia ołowiu w glebie jest zdeterminowana delikatną równowagą przeciwnie skierowanych procesów: sorpcja-desorpcja, rozpuszczanie-przejście do stanu stałego. Ołów uwalniany do gleby wraz z emisją włączany jest w cykl przemian fizycznych, chemicznych i fizykochemicznych. Początkowo dominują procesy mechanicznego przemieszczenia (cząstki ołowiu przemieszczają się po powierzchni iw glebie wzdłuż pęknięć) oraz dyfuzji konwekcyjnej. Następnie, w miarę rozpuszczania się związków ołowiu w fazie stałej, wchodzą w grę bardziej złożone procesy fizykochemiczne (w szczególności procesy dyfuzji jonów), którym towarzyszą przemiany związków ołowiu, które pochodzą z pyłu.
Ustalono, że ołów migruje zarówno pionowo, jak i poziomo, przy czym drugi proces dominuje nad pierwszym. W ciągu 3 lat obserwacji na łące pospolitej pył ołowiany aplikowany lokalnie na powierzchnię gleby przemieszczał się w kierunku poziomym o 25–35 cm, a głębokość jego wnikania w miąższość gleby wynosiła 10–15 cm.Ważną rolę odgrywają czynniki biologiczne migracja ołowiu: korzenie roślin absorbują jony metali; w okresie wegetacji poruszają się w grubości gleby; Kiedy rośliny obumierają i rozkładają się, ołów jest uwalniany do otaczającej masy gleby.
Wiadomo, że gleba ma zdolność wiązania (sorbcji) ołowiu technogenicznego, który do niej wszedł. Uważa się, że sorpcja obejmuje kilka procesów: całkowitą wymianę z kationami kompleksu absorbującego gleb (adsorpcja niespecyficzna) oraz szereg reakcji kompleksowania ołowiu z donorami składników glebowych (adsorpcja specyficzna). W glebie ołów kojarzy się głównie z materią organiczną, a także z minerałami ilastymi, tlenkami manganu, wodorotlenkami żelaza i glinu. Wiążąc ołów humus zapobiega jego migracji do sąsiednich środowisk i ogranicza jego przenikanie do roślin. Spośród minerałów ilastych illity charakteryzują się tendencją do sorpcji ołowiu. Wzrost pH gleby podczas wapnowania prowadzi do jeszcze większego wiązania ołowiu z glebą dzięki powstawaniu trudno rozpuszczalnych związków (wodorotlenki, węglany itp.).
Ołów, który jest obecny w glebie w postaci mobilnej, jest z czasem utrwalany przez składniki gleby i staje się niedostępny dla roślin. Według krajowych badaczy ołów jest najsilniej związany z czarnoziemami i glebami torfowo-mułowymi.
Kadm (Cd) Cechą odróżniającą kadm od innych HMs jest to, że występuje w roztworze glebowym głównie w postaci kationów (Cd 2+), chociaż w glebie o obojętnym odczynie środowiska może tworzyć trudno rozpuszczalne kompleksy z siarczanami, fosforanami lub wodorotlenkami.
Według dostępnych danych stężenie kadmu w roztworach glebowych gleb tła wynosi od 0,2 do 6 µg/l. W ogniskach zanieczyszczenia gleby wzrasta do 300-400 µg/l.
Wiadomo, że kadm w glebie jest bardzo mobilny; jest w stanie przejść w dużych ilościach z fazy stałej do cieczy i odwrotnie (co utrudnia przewidzenie jej wejścia do rośliny). Mechanizmy regulujące stężenie kadmu w roztworze glebowym determinowane są przez procesy sorpcji (przez sorpcję rozumiemy adsorpcję, opady i tworzenie kompleksów). Kadm jest wchłaniany przez glebę w mniejszych ilościach niż inne HM. Do scharakteryzowania ruchliwości metali ciężkich w glebie wykorzystuje się stosunek stężeń metali w fazie stałej do stężeń w roztworze równowagowym. Wysokie wartości tego stosunku wskazują, że HMs są zatrzymywane w fazie stałej na skutek reakcji sorpcji, niskie wartości - ze względu na fakt, że metale znajdują się w roztworze, skąd mogą migrować do innych mediów lub wnikać do różnych reakcje (geochemiczne lub biologiczne). Wiadomo, że wiodącym procesem w wiązaniu kadmu jest adsorpcja przez glinki. Ostatnie badania wykazały również dużą rolę w tym procesie grup hydroksylowych, tlenków żelaza i materii organicznej. Przy niskim poziomie zanieczyszczenia i obojętnym odczynie medium kadm jest adsorbowany głównie przez tlenki żelaza. A w środowisku kwaśnym (pH = 5) materia organiczna zaczyna działać jak silny adsorbent. Przy niższym pH (pH=4) funkcje adsorpcyjne przechodzą prawie wyłącznie na materię organiczną. Składniki mineralne w tych procesach przestają odgrywać jakąkolwiek rolę.
Wiadomo, że kadm jest nie tylko wchłaniany przez powierzchnię gleby, ale także utrwalany w wyniku opadów atmosferycznych, koagulacji i wchłaniania międzypakietów przez minerały ilaste. Dyfunduje do cząstek gleby przez mikropory i w inny sposób.
Kadm jest w różny sposób utrwalany w glebie inny rodzaj. Jak dotąd niewiele wiadomo o konkurencyjnych związkach kadmu z innymi metalami w procesach sorpcji w kompleksie glebochłonnym. Według badań przeprowadzonych przez specjalistów z Uniwersytetu Technicznego w Kopenhadze (Dania), w obecności niklu, kobaltu i cynku wchłanianie kadmu przez glebę ulegało zahamowaniu. Inne badania wykazały, że procesy sorpcji kadmu przez glebę zanikają w obecności jonów chlorkowych. Nasycenie gleby jonami Ca 2+ doprowadziło do zwiększenia pojemności sorpcyjnej kadmu. Wiele wiązań kadmu ze składnikami gleby okazuje się być kruchych, w pewnych warunkach (np. kwaśna reakcja środowiska) jest uwalniany i wraca do roztworu.
Ujawniono rolę drobnoustrojów w procesie rozpuszczania kadmu i jego przechodzenia do stanu ruchomego. W wyniku ich aktywności życiowej powstają albo rozpuszczalne w wodzie kompleksy metali, albo powstają warunki fizykochemiczne sprzyjające przechodzeniu kadmu z fazy stałej do ciekłej.
Procesy zachodzące z kadmem w glebie (sorpcja-desorpcja, przejście do roztworu itp.) są ze sobą powiązane i współzależne, przepływ tego metalu do roślin zależy od ich kierunku, intensywności i głębokości. Wiadomo, że wartość sorpcji kadmu przez glebę zależy od wartości pH: im wyższe pH gleby, tym więcej kadmu pochłania. Tak więc, według dostępnych danych, w zakresie pH od 4 do 7,7, wraz ze wzrostem pH na jednostkę, pojemność sorpcyjna gleb względem kadmu wzrosła około trzykrotnie.
Cynk (Zn). Niedobór cynku może objawiać się zarówno na glebach kwaśnych, silnie bielicowych lekkich, jak i węglanowych, ubogich w cynk i silnie próchnicznych. Przejaw niedoboru cynku potęguje stosowanie wysokich dawek nawozów fosforowych oraz silna orka podglebia do poziomu uprawnego.
Najwyższa zawartość cynku ogółem występuje w glebach tundry (53-76 mg/kg) i czarnoziemu (24-90 mg/kg), najmniejsza w glebach bielicowych (20-67 mg/kg). Niedobór cynku objawia się najczęściej w glebach wapiennych obojętnych i lekko zasadowych. W glebach kwaśnych cynk jest bardziej mobilny i dostępny dla roślin.
Cynk występuje w glebie w formie jonowej, gdzie jest adsorbowany na mechanizmie wymiany kationów w środowisku kwaśnym lub w wyniku chemisorpcji w środowisku zasadowym. Jon Zn 2+ jest najbardziej mobilny. Na mobilność cynku w glebie wpływa przede wszystkim wartość pH oraz zawartość minerałów ilastych. Przy pH<6 подвижность Zn 2+ возрастает, что приводит к его выщелачиванию. Попадая в межпакетные пространства кристаллической решетки монтмориллонита, ионы цинка теряют свою подвижность. Кроме того, цинк образует устойчивые формы с органическим веществом почвы, поэтому он накапливается в основном в горизонтах почв с высоким содержанием гумуса и в торфе.
Metale ciężkie w roślinach
Według A.P. Vinogradova (1952) wszystkie pierwiastki chemiczne biorą udział w życiu roślin w takim czy innym stopniu, a jeśli wiele z nich uważa się za fizjologicznie znaczące, to tylko dlatego, że nie ma na to jeszcze dowodów. Wchodząc do roślin w niewielkiej ilości i stając się integralną częścią lub aktywatorami zawartych w nich enzymów, mikroelement pełni funkcje usługowe w procesach metabolicznych. Kiedy niezwykle wysokie stężenia pierwiastków dostają się do środowiska, stają się toksyczne dla roślin. Wnikanie metali ciężkich do tkanek roślinnych w nadmiernych ilościach prowadzi do zakłócenia normalnego funkcjonowania ich narządów, a zakłócenie to jest tym silniejsze, im większy jest nadmiar toksyn. W rezultacie spada wydajność. Toksyczne działanie HM objawia się od wczesnych etapów rozwoju roślin, ale w różnym stopniu na różnych glebach i dla różnych upraw.
Przyswajanie pierwiastków chemicznych przez rośliny jest procesem aktywnym. Bierna dyfuzja to zaledwie 2-3% całkowitej masy trawionych składników mineralnych. Gdy zawartość metali w glebie jest na poziomie tła, następuje aktywna absorpcja jonów, a jeśli weźmiemy pod uwagę niską mobilność tych pierwiastków w glebie, to ich absorpcja powinna być poprzedzona mobilizacją silnie związanych metali. Gdy zawartość HM w warstwie korzeniowej jest w ilościach znacznie przekraczających graniczne stężenia, przy których metal może być utrwalony kosztem wewnętrznych zasobów gleby, do korzeni dostają się takie ilości metali, że błony nie są już w stanie zatrzymać. W efekcie podaż jonów czy związków pierwiastków przestaje być regulowana przez mechanizmy komórkowe. HM gromadzą się intensywniej na glebach kwaśnych niż na glebach o obojętnym lub bliskim obojętnym odczynie środowiska. Miarą rzeczywistego udziału jonów HM w reakcjach chemicznych jest ich aktywność. Toksyczny wpływ wysokich stężeń HM na rośliny może objawiać się zakłóceniami dostaw i dystrybucji innych pierwiastków chemicznych. Charakter oddziaływania HM z innymi pierwiastkami różni się w zależności od ich stężeń. Migracja i wejście do rośliny odbywa się w postaci złożonych związków.
W początkowym okresie zanieczyszczenia środowiska metalami ciężkimi, ze względu na buforowe właściwości gleby, prowadzące do inaktywacji toksyn, rośliny praktycznie nie odczują negatywnych skutków. Jednak funkcje ochronne gleby nie są nieograniczone. Wraz ze wzrostem poziomu zanieczyszczenia metalami ciężkimi ich dezaktywacja staje się niepełna, a strumień jonów atakuje korzenie. Część jonów roślina jest w stanie przejść w stan mniej aktywny jeszcze zanim przenikną one do systemu korzeniowego roślin. Jest to na przykład chelatacja za pomocą wydzieliny korzeniowej lub adsorpcja na zewnętrznej powierzchni korzeni z tworzeniem związków kompleksowych. Ponadto, jak wykazały doświadczenia wegetacyjne z oczywiście toksycznymi dawkami cynku, niklu, kadmu, kobaltu, miedzi i ołowiu, korzenie znajdują się w warstwach niezanieczyszczonych glebami HM i nie ma w tych wariantach objawów fototoksyczności.
Pomimo ochronnych funkcji systemu korzeniowego HMs wnikają do korzeni w warunkach zanieczyszczenia. W tym przypadku w grę wchodzą mechanizmy ochronne, dzięki którym następuje specyficzne rozmieszczenie HMs pomiędzy organami roślin, co pozwala na możliwie najpełniejsze zabezpieczenie ich wzrostu i rozwoju. Jednocześnie zawartość np. HMs w tkankach korzeni i nasion w warunkach silnie zanieczyszczonego środowiska może różnić się od 500 do 600 razy, co wskazuje na duże zdolności ochronne tego podziemnego organu roślinnego.
Nadmiar pierwiastków chemicznych powoduje zatrucie roślin. Wraz ze wzrostem stężenia HM wzrost roślin jest początkowo opóźniony, następnie rozpoczyna się chloroza liści, która zostaje zastąpiona przez nekrozę, aw końcu dochodzi do uszkodzenia systemu korzeniowego. Toksyczny efekt HM może objawiać się bezpośrednio i pośrednio. Bezpośredni wpływ nadmiaru HM w komórkach roślinnych wynika z reakcji tworzenia kompleksów, w wyniku których dochodzi do blokowania enzymów lub wytrącania białek. Dezaktywacja układów enzymatycznych następuje w wyniku zastąpienia metalu enzymatycznego zanieczyszczeniem metalowym. Przy krytycznej zawartości substancji toksycznej zdolność katalityczna enzymu jest znacznie zmniejszona lub całkowicie zablokowana.
Rośliny są hiperakumulatorami metali ciężkich
AP Vinogradov (1952) wyróżnił rośliny zdolne do koncentracji pierwiastków. Wskazał na dwa rodzaje roślin - koncentratory: 1) rośliny koncentrujące pierwiastki na masową skalę; 2) rośliny o koncentracji selektywnej (gatunkowej). Rośliny pierwszego typu wzbogaca się w pierwiastki chemiczne, jeśli te ostatnie są zawarte w glebie w zwiększonej ilości. Stężenie w tym przypadku jest spowodowane czynnikiem środowiskowym. Rośliny drugiego typu charakteryzują się stale dużą ilością tego czy innego pierwiastka chemicznego, niezależnie od jego zawartości w środowisku. Wynika to z uwarunkowanej genetycznie potrzeby.
Rozważając mechanizm wchłaniania metali ciężkich z gleby do roślin, możemy mówić o barierowych (niekoncentrujących) i bezbarierowych (koncentrujących) typach akumulacji pierwiastków. Akumulacja barierowa jest charakterystyczna dla większości roślin wyższych i nie jest charakterystyczna dla mszaków i porostów. Tak więc w pracy M. A. Toikki i L. N. Potekhiny (1980) torfowiec (2,66 mg/kg) został nazwany roślinnym koncentratorem kobaltu; miedź (10,0 mg/kg) - brzoza, owoce pestkowe, konwalia; mangan (1100 mg/kg) - jagody. Lepp i in. (1987) stwierdzili wysokie stężenia kadmu w sporoforach grzyba Amanita muscaria rosnącego w lasach brzozowych. W sporoforach grzyba zawartość kadmu wynosiła 29,9 mg/kg sm, aw glebie, na której rosły, 0,4 mg/kg. Istnieje opinia, że rośliny będące koncentratorami kobaltu są również bardzo tolerancyjne na nikiel i potrafią go akumulować w dużych ilościach. Należą do nich w szczególności rośliny z rodzin Boraginaceae, Brassicaceae, Myrtaceae, Fabaceae, Caryophyllaceae. Wśród roślin leczniczych znajdują się również koncentratory i superkoncentratory niklu. Do superkoncentratorów należą melon, belladonna belladonna, żółta mache, motherwort, mięsna męczennica i lancetowata termopsja. Rodzaj akumulacji pierwiastków chemicznych o wysokim stężeniu w pożywce zależy od faz wegetacji roślin. Akumulacja bezbarierowa jest typowa dla fazy siewek, kiedy rośliny nie mają zróżnicowania części nadziemnych na różne organy, a w końcowych fazach wegetacji - po dojrzewaniu, a także w okresie spoczynku zimowego, kiedy akumulacja bezbarierowa może towarzyszyć może uwalnianie nadmiernych ilości pierwiastków chemicznych w fazie stałej (Kovalevsky, 1991).
Rośliny hiperakumulujące znaleziono w rodzinach Brassicaceae, Euphorbiaceae, Asteraceae, Lamiaceae i Scrophulariaceae (Baker 1995). Najbardziej znaną i zbadaną wśród nich jest Brassica juncea (gorczyca indyjska) – roślina, która rozwija dużą biomasę i jest zdolna do akumulacji Pb, Cr (VI), Cd, Cu, Ni, Zn, 90Sr, B i Se (Nanda Kumar i wsp. 1995; Salt i wsp. 1995; Raskin i wsp. 1994). Spośród różnych badanych gatunków roślin B. juncea miała najsilniejszą zdolność przenoszenia ołowiu do części nadziemnych, gromadząc w organach nadziemnych ponad 1,8% tego pierwiastka (w przeliczeniu na suchą masę). Z wyjątkiem słonecznika (Helianthus annuus) i tytoniu (Nicotiana tabacum), inne gatunki roślin spoza rodziny Brassicaceae miały współczynnik biodostępności poniżej 1.
Zgodnie z klasyfikacją roślin według reakcji na obecność metali ciężkich w podłożu, stosowaną przez wielu autorów zagranicznych, rośliny mają trzy główne strategie uprawy na glebach zanieczyszczonych metalami:
Metalowe odgrody. Takie rośliny zachowują stałą, niską koncentrację tego metalu pomimo dużej zmienności jego stężenia w glebie, zatrzymując głównie metal w korzeniach. Rośliny wykluczające są zdolne do zmiany przepuszczalności błony i zdolności ścian komórkowych do wiązania metali lub uwalniania dużych ilości środków chelatujących.
Wskaźniki metalowe. Należą do nich gatunki roślin, które aktywnie gromadzą metal w częściach nadziemnych i ogólnie odzwierciedlają poziom zawartości metalu w glebie. Są tolerancyjne na obecny poziom stężenia metali ze względu na tworzenie się pozakomórkowych związków wiążących metale (chelatory) lub zmieniają charakter przedziałów metali poprzez przechowywanie ich w obszarach niewrażliwych na metal. Gatunki roślin gromadzące metale. Rośliny należące do tej grupy mogą akumulować metal w biomasie nadziemnej w stężeniach znacznie wyższych niż w glebie. Baker i Brooks zdefiniowali hiperakumulatory metali jako rośliny zawierające więcej niż 0,1%, tj. ponad 1000 mg/g miedzi, kadmu, chromu, ołowiu, niklu, kobaltu lub 1% (ponad 10 000 mg/g) cynku i manganu w suchej masie. W przypadku metali rzadkich wartość ta przekracza 0,01% suchej masy. Naukowcy identyfikują gatunki hiperakumulacyjne, zbierając rośliny z obszarów, na których gleba zawiera metale w stężeniach przekraczających poziomy tła, na przykład na obszarach skażonych lub wychodniach rudy. Zjawisko hiperakumulacji rodzi wiele pytań badaczom. Na przykład, jakie jest znaczenie akumulacji metali w wysoce toksycznych stężeniach dla roślin. Nie otrzymano jeszcze ostatecznej odpowiedzi na to pytanie, ale istnieje kilka głównych hipotez. Uważa się, że takie rośliny mają wzmocniony system pobierania jonów (hipoteza „niezamierzonego” pobierania) do wykonywania pewnych funkcji fizjologicznych, które nie zostały jeszcze zbadane. Uważa się również, że hiperakumulacja jest jednym z rodzajów tolerancji roślin na wysoką zawartość metali w środowisku uprawy.
Metale ciężkie (HM) obejmują ponad 40 pierwiastków chemicznych układu okresowego D. I. Mendelejewa, których masa atomów wynosi ponad 50 jednostek masy atomowej (amu). Są to Pb, Zn, Cd, Hg, Cu, Mo, Mn, Ni, Sn, Co itp.
Obecna koncepcja „metali ciężkich” nie jest ścisła, ponieważ pierwiastki niemetaliczne, takie jak As, Se, a czasem nawet F, Be i inne pierwiastki, których masa atomowa jest mniejsza niż 50 am, są często określane jako HM.
Wśród HM znajduje się wiele pierwiastków śladowych, które są ważne biologicznie dla żywych organizmów. Są niezbędnymi i niezastąpionymi składnikami biokatalizatorów i bioregulatorów najważniejszych procesów fizjologicznych. Jednak nadmierna zawartość HMs w różnych obiektach biosfery działa przygnębiająco, a nawet toksycznie na organizmy żywe.
Źródła HM przedostające się do gleby dzielą się na naturalne (wietrzenie skał i minerałów, procesy erozyjne, aktywność wulkaniczna) i technogeniczne (wydobycie i przetwarzanie minerałów, spalanie paliw, oddziaływanie pojazdów, rolnictwo itp.) Użytki rolne, dodatkowo do zanieczyszczenia z atmosfery, są również szczególnie zanieczyszczone HM w przypadku stosowania pestycydów, nawozów mineralnych i organicznych, wapnowania i korzystania ze ścieków. Ostatnio naukowcy zwrócili szczególną uwagę na gleby miejskie. Te ostatnie doświadczają znacznego ciśnienia technogenicznego, którego integralną częścią są zanieczyszczenia HM.
W tabeli. Rysunki 3.14 i 3.15 przedstawiają rozmieszczenie HM w różnych obiektach biosfery oraz źródła przedostawania się HM do środowiska.
Tabela 3.14
| Element | Gleby | świeża woda | wody morskie | Rośliny | Zwierzęta (w tkance mięśniowej) |
| Mn | 1000 | 0,008 | 0,0002 | 0,3-1000 | 0,2-2,3 |
| Zn | 90 (1-900) | 0,015 | 0,0049 | 1,4-600 | 240 |
| Cu | 30 (2-250) | 0,003 | 0,00025 | 4-25 | 10 |
| współ | 8 (0,05-65) | 0,0002 | 0,00002 | 0,01-4,6 | 0,005-1 |
| Pb | 35 (2-300) | 0,003 | 0,00003 | 0,2-20 | 0,23-3,3 |
| płyta CD | 0,35 (0,01-2) | 0,0001 | - | 0,05-0,9 | 0,14-3,2 |
| hg | 0,06 | 0,0001 | 0,00003 | 0,005-0,02 | 0,02-0,7 |
| Jak | 6 | 0,0005 | 0,0037 | 0,02-7 | 0,007-0,09 |
| Se | 0,4 (0,01-12) | 0,0002 | 00,0002 | 0,001-0,5 | 0,42-1,9 |
| F | 200 | 0,1 | 1,3 | 0,02-24 | 0,05 |
| B | 20 (2-270) | 0,15 | 4,44 | 8-200 | 0,33-1 |
| Mo | 1,2 (0,1-40) | 0,0005 | 0,01 | 0,03-5 | 0,02-0,07 |
| Cr | 70 (5-1500) | 0,001 | 0,0003 | 0,016-14 | 0,002-0,84 |
| Ni | 50 (2-750) | 0,0005 | 0,00058 | 0,02-4 | 1-2 |
Tabela 3.15
Źródła zanieczyszczenia środowiska HM
Koniec tabeli. 3.4
HMs docierają do powierzchni gleby w różnych formach. Są to tlenki i różne sole metali, zarówno rozpuszczalne, jak i praktycznie nierozpuszczalne w wodzie (siarczki, siarczany, arseniny itp.). W składzie emisji z zakładów przetwórstwa rud i zakładów metalurgii metali nieżelaznych - głównego źródła zanieczyszczenia środowiska HM - większość metali (70-90%) występuje w postaci tlenków.
Dostając się na powierzchnię gleby, HM mogą albo gromadzić się, albo rozpraszać, w zależności od charakteru barier geochemicznych właściwych dla danego terytorium.
Większość HM, które dostały się do powierzchni gleby, jest utrwalona w górnych warstwach próchniczych. HMs są sorbowane na powierzchni cząstek gleby, wiążą się z materią organiczną gleby, w szczególności w postaci pierwiastkowych związków organicznych, gromadzą się w wodorotlenkach żelaza, wchodzą w skład sieci krystalicznych minerałów ilastych, dają własne minerały w wyniku izomorfii substytucji i są w stanie rozpuszczalnym w wilgotności gleby oraz w stanie gazowym w powietrzu glebowym, stanowią integralną część bioty glebowej.
Stopień ruchliwości HM zależy od środowiska geochemicznego i poziomu oddziaływania technogenicznego. Duży rozkład wielkości cząstek i wysoka zawartość materii organicznej prowadzą do wiązania HMs przez glebę. Wzrost wartości pH wzmaga sorpcję metali kationotwórczych (miedź, cynk, nikiel, rtęć, ołów itp.) oraz zwiększa mobilność metali anionotwórczych (molibden, chrom, wanad itp.). Wzmocnienie warunków utleniających zwiększa zdolność migracji metali. W efekcie, zgodnie ze zdolnością do wiązania większości HMs, gleby tworzą następujące serie: gleba szara > czarnoziem > gleba bagienno-bielicowa.
Czas przebywania składników zanieczyszczających w glebie jest znacznie dłuższy niż w innych częściach biosfery, a zanieczyszczenie gleby, zwłaszcza HM, jest praktycznie wieczne. Metale gromadzące się w glebie są powoli usuwane przez ługowanie, konsumpcję przez rośliny, erozję i deflację (Kabata-Pendias, Pendias, 1989). Okres połowicznego usunięcia (lub usunięcia połowy początkowego stężenia) HM jest bardzo zróżnicowany dla różnych pierwiastków, ale jest dość długi: dla Zn - od 70 do 510 lat; dla Cd - od 13 do 110 lat; dla Cu - od 310 do 1500 lat, a dla Pb - 2 - od 740 do 5900 lat (Sadovskaya, 1994).
Zanieczyszczenie gleby HM ma jednocześnie dwie negatywne strony. Po pierwsze, HM, przechodząc przez łańcuchy pokarmowe z gleby do roślin, a stamtąd do organizmu zwierząt i ludzi, powodują u nich poważne choroby - wzrost zachorowalności populacji i skrócenie oczekiwanej długości życia, a także spadek w ilości i jakości plonów roślin rolniczych i produktów zwierzęcych.
Po drugie, gromadząc się w glebie w dużych ilościach, HMs mogą zmieniać wiele jej właściwości. Przede wszystkim zmiany wpływają na biologiczne właściwości gleby: zmniejsza się całkowita liczba mikroorganizmów, zawęża się ich skład gatunkowy (różnorodność), zmienia się struktura zbiorowisk mikroorganizmów, intensywność głównych procesy mikrobiologiczne i aktywność enzymów glebowych itp. Silne zanieczyszczenie HM prowadzi również do zmian w bardziej konserwatywnych cechach gleby, takich jak stan próchnicy, struktura, pH itp. Powoduje to częściową, a w niektórych przypadkach całkowitą utratę żyzność gleby.
W przyrodzie występują obszary o niedostatecznej lub nadmiernej zawartości HM w glebach. Anomalna zawartość HM w glebach wynika z dwóch grup przyczyn: biogeochemicznych cech ekosystemów oraz wpływu technogenicznych strumieni materii. W pierwszym przypadku obszary, w których stężenie pierwiastków chemicznych jest powyżej lub poniżej poziomu optymalnego dla organizmów żywych, nazywane są naturalnymi anomaliami geochemicznymi lub prowincjami biogeochemicznymi. Tutaj anomalna zawartość pierwiastków wynika z przyczyn naturalnych - cech skał glebotwórczych, procesu glebotwórczego, obecności anomalii kruszcowych. W drugim przypadku terytoria nazywane są technogenicznymi anomaliami geochemicznymi. W zależności od skali dzielą się na globalne, regionalne i lokalne.
Gleba, w przeciwieństwie do innych składników środowiska przyrodniczego, nie tylko geochemicznie akumuluje składniki zanieczyszczeń, ale pełni również rolę naturalnego bufora kontrolującego przenikanie pierwiastków i związków chemicznych do atmosfery, hydrosfery i materii żywej.
Różne rośliny, zwierzęta i ludzie wymagają do życia określonego składu gleby i wody. W miejscach anomalii geochemicznych następuje, nasilone, przenoszenie odchyleń od normy składu mineralnego w całym łańcuchu pokarmowym.
W wyniku naruszenia żywienia mineralnego, zmian w składzie gatunkowym cenoz fito-, zoo- i mikrobiologicznych, chorób dziko rosnących form roślin, spadku ilości i jakości plonów roślin rolniczych i produktów zwierzęcych, obserwuje się wzrost zapadalności w populacji i skrócenie oczekiwanej długości życia (tab. 3.15). Mechanizm działania toksycznego HM przedstawiono w tabeli. 3.16.
Tabela 3.15
Zaburzenia fizjologiczne w roślinach z nadmiarem i niedoborem zawartości HM w nich (wg Kovalevsky i Andrianova, 1970; Kabata-pendias,
pendia, 1989)
| Element | Zaburzenia fizjologiczne | |
| z brakiem | w nadmiarze | |
| Cu | Chloroza, więdnięcie, melanizm, białe poskręcane wierzchołki, zmniejszone tworzenie wiech, upośledzone zdrewnienie, martwe wierzchołki drzew | Liście ciemnozielone, jak w chlorozie indukowanej Fe; grube, krótkie lub kolczaste korzenie przypominające drut, zahamowanie tworzenia pędów |
| Zn | Chloroza międzynerwowa (głównie u jednoliściennych), zahamowanie wzrostu, rozeta liści drzew, fioletowo-czerwone kropki na liściach | Chloroza i nekroza końcówek liści, międzynerwowa chloroza młodych liści, karłowatość rośliny jako całości, uszkodzone korzenie przypominające drut kolczasty |
| płyta CD | - | Brązowe brzegi liści, chloroza, czerwonawe żyłki i ogonki liściowe, skręcone liście i niedorozwinięte brązowe korzenie |
| hg | - | Pewne zahamowanie pędów i korzeni, chloroza liści i brązowe plamy na nich |
| Pb | - | Zmniejszone tempo fotosyntezy, ciemnozielone liście, zwijanie się starych liści, karłowate liście, krótkie brązowe korzenie |
Tabela 3.16
Mechanizm działania toksyczności HM (wg Torshina i wsp., 1990)
| Element | Akcja |
| Cu, Zn, Cd, Hg, Pb | Wpływ na przepuszczalność błony, reakcja z SH - grupami cysteiny i metioniny |
| Pb | Zmiana trójwymiarowej struktury białek |
| Cu, Zn, Hg, Ni | Tworzenie kompleksów z fosfolipidami |
| Ni | Tworzenie kompleksów z albuminami |
| Hamowanie enzymów: | |
| Hg2+ | fosfataza alkaliczna, gluko-6-fosfataza, dehydrogenaza mleczanowa |
| CD2+ | trifosfataza adenozynowa, dehydrogenaza alkoholowa, amylaza, anhydraza węglanowa, karboksypeptydazy (pentidazy), transaminazy glutamatoksalooctanowe |
| Pb2+ | acetylocholinoesteraza, fosfataza alkaliczna, ATPaza |
| Ni2+ | anhydraza węglanowa, oksydaza cytochromowa, hydroksylaza benzopirenowa |
Toksyczny wpływ HM na systemy biologiczne przede wszystkim ze względu na to, że łatwo wiążą się z grupami sulfhydrylowymi białek (w tym enzymów), hamując ich syntezę i tym samym zaburzając metabolizm organizmu.
Żywe organizmy wykształciły różne mechanizmy odporności na HM: od redukcji jonów HM do mniej toksycznych związków po aktywację systemów transportu jonów, które skutecznie i specyficznie usuwają toksyczne jony z komórki do środowiska zewnętrznego.
Najważniejszą konsekwencją oddziaływania HM na organizmy żywe, przejawiającą się na biogeocenotycznym i biosferycznym poziomie organizacji materii żywej, jest blokowanie procesów utleniania materii organicznej. Prowadzi to do zmniejszenia tempa jego mineralizacji i akumulacji w ekosystemach. Jednocześnie wzrost stężenia materii organicznej powoduje wiązanie HMs, co czasowo odciąża ekosystem. Spadek tempa rozkładu materii organicznej w wyniku zmniejszenia liczby organizmów, ich biomasy i intensywności aktywności życiowej jest uważany za bierną reakcję ekosystemów na zanieczyszczenie HM. Aktywny opór organizmów wobec obciążeń antropogenicznych przejawia się tylko podczas dożywotniego nagromadzenia metali w ciałach i szkieletach. Za ten proces odpowiadają najbardziej odporne gatunki.
Odporność organizmów żywych, przede wszystkim roślin, na podwyższone stężenia HM i ich zdolność do akumulacji wysokich stężeń metali może stanowić duże zagrożenie dla zdrowia ludzkiego, ponieważ umożliwiają przenikanie zanieczyszczeń do łańcuchów pokarmowych. W zależności od geochemicznych warunków produkcji, żywność ludzka, zarówno pochodzenia roślinnego, jak i zwierzęcego, może zaspokajać ludzkie zapotrzebowanie na pierwiastki mineralne, może być niedoborowa lub zawierać ich nadmiar, stając się bardziej toksyczna, powodując choroby, a nawet śmierć (tab. 3.17).
Tabela 3.17
Wpływ HM na organizm ludzki (Kowalsky, 1974; Brief Medical Encyclopedia, 1989; Torshin i in., 1990; Effects on the body.., 1997; Handbook of toxicology.., 1999)
| Element | Nieprawidłowości fizjologiczne | |
| z brakiem | w nadmiarze | |
| Mn | Choroby układu kostnego | Gorączka, zapalenie płuc, uszkodzenie ośrodkowego układu nerwowego (parkinsonizm manganowy), endemiczna dna moczanowa, zaburzenia krążenia, funkcje przewodu pokarmowego, niepłodność |
| Cu | Osłabienie, anemia, białaczka, choroby układu kostnego, zaburzona koordynacja ruchów | Choroby zawodowe, zapalenie wątroby, choroba Wilsona. Wpływa na nerki, wątrobę, mózg, oczy |
| Zn | Zmniejszony apetyt, deformacja kości, wzrost karłowaty, długie gojenie się ran i oparzeń, słaby wzrok, krótkowzroczność | Zmniejszona odporność na nowotwory, anemia, zahamowanie procesów oksydacyjnych, zapalenie skóry |
| Pb | - | Encefaloneuropatia ołowiowa, zaburzenia metaboliczne, hamowanie reakcji enzymatycznych, beri-beri, niedokrwistość, stwardnienie rozsiane. Zawarty w układzie kostnym zamiast wapnia |
| płyta CD | - | Zaburzenia żołądka i jelit, zaburzenia oddechowe, anemia, wysokie ciśnienie krwi, uszkodzenie nerek, choroba itai-itai, białkomocz, osteoporoza, działanie mutagenne i rakotwórcze |
| hg | - | Uszkodzenia ośrodkowego układu nerwowego i nerwów obwodowych, infantylizm, upośledzenie funkcji rozrodczych, zapalenie jamy ustnej, choroby Minamata, przedwczesne starzenie |
| współ | wole endemiczne | - |
| Ni | - | Zapalenie skóry, zaburzenia hematopoetyczne, rakotwórczość, embriotoksyczne, podostra neuropatia mielo-wzrokowa |
| Cr | - | Zapalenie skóry, rakotwórczość |
| V | - | Choroby układu sercowo-naczyniowego |
Różne HM w różnym stopniu zagrażają zdrowiu ludzkiemu. Najbardziej niebezpieczne są Hg, Cd, Pb (tabela 3.18).
Tabela 3.18
Klasy zanieczyszczeń według stopnia zagrożenia (GOST 17.4.1.02-83)
Kwestia reglamentacji zawartości HMs w glebie jest bardzo skomplikowana. Podstawą jego decyzji powinno być uznanie wielofunkcyjności gleby. W procesie racjonowania glebę można rozpatrywać z różnych pozycji: jako ciało naturalne; jako siedlisko i podłoże dla roślin, zwierząt i mikroorganizmów; jako przedmiot i środek rolnictwa i produkcja przemysłowa; jako naturalny zbiornik zawierający drobnoustroje chorobotwórcze. Racjonowanie zawartości HMs w glebie powinno być prowadzone w oparciu o zasady glebo-ekologiczne, które negują możliwość znalezienia jednolitych wartości dla wszystkich gleb.
Istnieją dwa główne podejścia do kwestii sanitacji gleb skażonych HM. Pierwsza ma na celu oczyszczenie gleby z HM. Oczyszczanie można przeprowadzić przez płukanie, ekstrakcję HM z gleby za pomocą roślin, usunięcie wierzchniej, skażonej warstwy gleby itp. Drugie podejście opiera się na utrwalaniu HM w glebie, przekształcaniu ich w formy nierozpuszczalne w wodzie i niedostępne dla żywych organizmów. W tym celu proponuje się wprowadzenie do gleby materii organicznej, fosforowych nawozów mineralnych, żywic jonowymiennych, naturalnych zeolitów, węgla brunatnego, wapnowania gleby itp. Jednak każda metoda wiązania HM w glebie ma swój okres ważność. Prędzej czy później część HM ponownie zacznie wnikać do roztworu glebowego, a stamtąd do żywych organizmów.
Tak więc ponad 40 pierwiastków chemicznych jest klasyfikowanych jako metale ciężkie, których masa atomów przekracza 50 amu. jeść. Są to Pb, Zn, Cd, Hg, Cu, Mo, Mn, Ni, Sn, Co itp. Wśród HMs znajduje się wiele pierwiastków śladowych, które są niezbędnymi i niezastąpionymi składnikami biokatalizatorów i bioregulatorów najważniejszych procesów fizjologicznych. Jednak nadmierna zawartość HMs w różnych obiektach biosfery działa przygnębiająco, a nawet toksycznie na organizmy żywe.
Źródła wnikania HM do gleby dzielą się na naturalne (wietrzenie skał i minerałów, procesy erozyjne, aktywność wulkaniczna) oraz technogeniczne (wydobycie i przetwarzanie minerałów, spalanie paliw, oddziaływanie pojazdów, rolnictwa itp.).
HMs docierają do powierzchni gleby w różnych formach. Są to tlenki i sole różnych metali, zarówno rozpuszczalne, jak i praktycznie nierozpuszczalne w wodzie.
Ekologiczne konsekwencje zanieczyszczenia gleby HM zależą od parametrów zanieczyszczenia, warunków geochemicznych i stabilności gleby. Parametry zanieczyszczenia obejmują rodzaj metalu, tj. jego właściwości chemiczne i toksyczne, zawartość metalu w glebie, postać związku chemicznego, okres od momentu zanieczyszczenia itp. Odporność gleby na zanieczyszczenia zależy od wielkości cząstek rozmieszczenie, zawartość materii organicznej, warunki kwasowo-zasadowe i redoks, aktywność procesów mikrobiologicznych i biochemicznych itp.
Odporność organizmów żywych, przede wszystkim roślin, na podwyższone stężenia HM i ich zdolność do akumulacji wysokich stężeń metali może stanowić duże zagrożenie dla zdrowia ludzkiego, ponieważ umożliwiają przenikanie zanieczyszczeń do łańcuchów pokarmowych.
Normalizując zawartość HMs w glebie należy wziąć pod uwagę wielofunkcyjność gleby. Gleba może być traktowana jako organizm naturalny, siedlisko i podłoże dla roślin, zwierząt i mikroorganizmów, przedmiot i środek produkcji rolniczej i przemysłowej, naturalny zbiornik zawierający mikroorganizmy chorobotwórcze, element biogeocenozy lądowej i biosfery jako całość.